【生信分析】基因组学导论
由于工作需要,现在开始跨行学生信!
祝我成功
目标:通过一周的学习能对对不同高通量测序数据集(RNA-seq、ChIP-seq、BS-seq 和多组学集成)进行分析。
配置环境
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager") BiocManager::install(c('qvalue','plot3D','ggplot2','pheatmap','cowplot', 'cluster', 'NbClust', 'fastICA', 'NMF','matrixStats', 'Rtsne', 'mosaic', 'knitr', 'genomation', 'ggbio', 'Gviz', 'DESeq2', 'RUVSeq', 'gProfileR', 'ggfortify', 'corrplot', 'gage', 'EDASeq', 'citr', 'formatR', 'svglite', 'Rqc', 'ShortRead', 'QuasR', 'methylKit','FactoMineR', 'iClusterPlus', 'enrichR','caret','xgboost','glmnet', 'DALEX','kernlab','pROC','nnet','RANN', 'ranger','GenomeInfoDb', 'GenomicRanges', 'GenomicAlignments', 'ComplexHeatmap', 'circlize', 'rtracklayer', 'BSgenome.Hsapiens.UCSC.hg38', 'BSgenome.Hsapiens.UCSC.hg19','tidyr', 'AnnotationHub', 'GenomicFeatures', 'normr', 'MotifDb', 'TFBSTools', 'rGADEM', 'JASPAR2018' ))
第一章:“基因组学导论”
介绍基因组生物学和基因组学的基本概念。理解这些概念对于计算基因组学非常重要。
1.1 基因、DNA 和中心法则( Genes, DNA and central dogma )
一个不断出现的中心概念是“基因”。在我们解释这一点之前,我们需要介绍一些对于理解基因概念很重要的其他概念。人体由数十亿个细胞组成。这些细胞专门从事不同的任务。例如,肝脏中的细胞有助于产生酶来分解毒素。在心脏中,有专门的肌肉细胞使心脏跳动。然而,所有这些不同种类的细胞都来自单细胞胚胎。制造不同类型细胞的所有指令都包含在该单个细胞内,并且随着该细胞的每次分裂,这些指令都会传输到新细胞。这些指令可以被编码成一个字符串——DNA分子,一种由称为核苷酸的重复单元组成的聚合物。 DNA分子中的四种核苷酸,腺嘌呤、鸟嘌呤、胞嘧啶和胸腺嘧啶(编码为四个字母:A、C、G和T)以特定的顺序存储着生命的信息。 DNA 以双螺旋形式组织,其中两个互补的聚合物彼此交织并扭曲成熟悉的螺旋形状。
1.1.1 什么是基因组?(What is a genome?)
生物体的完整 DNA 序列包含所有遗传信息,称为基因组。基因组包含构建和维持有机体的所有信息。基因组有不同的大小和结构。我们的基因组不仅仅是一段裸露的DNA。在真核细胞中,DNA 包裹在蛋白质(组蛋白)周围,形成高级结构,如构成染色质和染色体的核小体(见图 1.1)。
根据生物体的不同,可能有几条染色体。然而,在某些物种(例如大多数原核生物)中,DNA 以环状形式储存。物种之间基因组的大小也不同。人类基因组有46条染色体和超过30亿个碱基对,而小麦基因组有42条染色体和170亿个碱基对;不同生物体之间的基因组大小和染色体数量都是可变的。使用测序技术获得生物体的基因组序列。通过这项技术,可以获得基因组中的 DNA 序列片段,称为“reads”。随后通过使用重叠读取将初始片段拼接到更大的片段来获得更大的基因组序列块。最新的测序技术使基因组测序变得更便宜、更快。这些技术输出更多的读数、更长的读数和更准确的读数。 1999-2000 年,第一个人类基因组的估计成本为 3 亿美元;如今,只需 1500 美元即可获得高质量的人类基因组。由于成本下降,研究人员和临床医生可以生成更多数据。这增加了数据存储的成本,也增加了对分析基因组数据的合格人员的需求。这是写这本书的动机之一。
1.1.2 什么是基因?
在基因组中,有一些特定区域包含编码遗传信息物理产物的精确信息。基因组中具有此信息的区域传统上称为“基因”。然而,该基因的精确定义仍在发展中。根据分子生物学的经典教科书,基因是对应于单个蛋白质或单个催化和结构RNA分子的DNA序列片段(Alberts等,2002)。现代定义是:“包含所有序列元素的区域(或多个区域)编码功能性转录本所必需的”(Eilbeck et al.,2005)。无论定义如何变化,所有人都同意这样一个事实:基因是所有生物体遗传的基本单位。
大多数时候,所有细胞都以相同的方式使用其遗传信息; DNA 被复制以将信息转移到新细胞。如果被激活,基因会在细胞核(真核生物)中转录成信使 RNA (mRNA),然后 mRNA(如果基因编码蛋白质)在细胞质中翻译成蛋白质。这本质上是承载信息的聚合物之间的信息传递过程; DNA、RNA和蛋白质,被称为分子生物学的“中心法则”(总结见图1.2)。蛋白质是生命必需的元素。所有活细胞的生长和修复、功能和结构都依赖于它们。这就是为什么基因是基因组生物学的中心概念,因为基因可以编码蛋白质和其他功能分子的信息。基因如何控制和激活决定了有机体的一切。从细胞的身份到对感染的反应,细胞如何发育以及针对某些刺激的行为是由基因及其编码的功能分子的活性控制的。肝细胞之所以成为肝细胞,是因为某些基因被激活,并产生它们的功能产物,帮助肝细胞完成其任务。
1.1.3 基因是如何控制的?
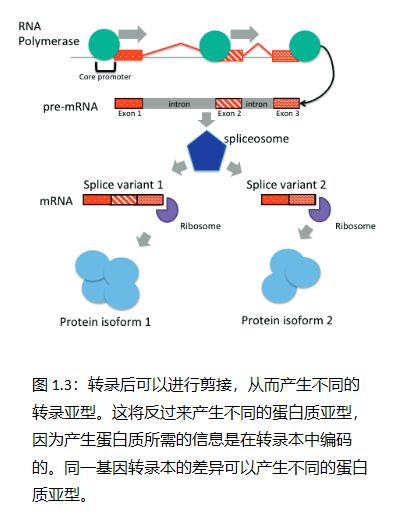
转录和转录后调控为了回答这个问题,我们必须更深入地研究我们通过中心法则引入的转录概念。信息传递过程的第一步——产生部分 DNA 序列的 RNA 副本——称为转录。这项任务是由 RNA 聚合酶完成的。 DNA 序列中特定区域(核心启动子)的存在使得 RNA 聚合酶依赖性转录起始成为可能。核心启动子是促进转录的 DNA 区域,位于转录起始位点的上游。在真核生物中,几种称为通用转录因子的蛋白质识别并结合核心启动子并形成预启动复合物。 RNA 聚合酶识别这些复合物并启动 RNA 的合成,聚合酶沿着模板 DNA 行进并生成 RNA 副本(Hager 等,2009)。 mRNA 产生后,通常通过剪接体进行剪接。这些被称为“内含子”的部分被移除,而被称为“外显子”的部分被保留。然后,剩余的 mRNA 被翻译成蛋白质。哪些外显子将成为最终成熟转录本的一部分也可以进行调节,并创造蛋白质结构和功能的多样性(见图 1.3)。
与蛋白质编码基因相反,非编码 RNA (ncRNA) 基因在转录后经过加工并呈现其功能结构,无需进行翻译,因此得名:非编码 RNA。某些 ncRNA 也可以被剪接,但仍然不能翻译。 ncRNA 和其他 RNA 通常可以在 RNA 分子内形成互补碱基对,这赋予它们额外的复杂性。这基于自我互补的结构,称为 RNA 二级结构,通常是许多 ncRNA 物种功能所必需的。总之,从转录起始到功能产物产生的一系列过程被称为基因表达。基因表达定量和调控是基因组生物学的一个基本主题。
1.1.4 基因是什么样的?
在我们继续前进之前,最好先讨论一下如何可视化基因。作为对计算基因组学感兴趣的人,您会经常在计算机屏幕上遇到一个基因,而它在计算机上的表示方式将等同于您听到“基因”这个词时所想象的。在在线数据库中,基因将显示为字母序列或一系列连接的框,显示外显子-内含子结构,其中也可能包括转录方向(见图 1.4)。你会遇到更多关于后者的情况,所以当你想到基因时,你可能会想到这一点。正如我们所提到的,DNA 有两条链。基因可以位于它们中的任何一个上,转录的方向将取决于该基因。在图 1.4 中,您可以看到内含子上的箭头(连接框的线)指示基因的方向。
1.2 基因调控的要素 (gene regulation)
调控基因表达的机制对于所有生物体都是至关重要的,因为它们决定了基因产物(可能是蛋白质或 ncRNA)的生产地点和数量。通过控制应产生多少转录本和/或应通过调节剪接产生哪个版本的转录本,这种调节可以发生在转录前和共转录水平。相同的基因可以通过剪接调节编码相同蛋白质的不同版本。这个过程定义了基因的哪些部分将进入最终的 mRNA,从而编码蛋白质变体。此外,基因产物可以在转录后进行调节,其中某些分子与RNA结合,甚至在它们用于蛋白质生产之前就标记它们以进行降解。基因调控驱动细胞分化;产生不同组织和细胞类型的过程。它还帮助细胞维持细胞/组织的分化状态。这一过程的结果是,在分化的最后阶段,不同种类的细胞尽管含有相同的遗传物质,但仍保持不同的表达谱。如上所述,监管主要有两种类型,接下来我们将提供这方面的信息。
1.2.1 转录调控 (Transcriptional regulation)
转录起始速率是基因表达调控的首要调控元件。该速率由核心启动子元件以及增强子等远距离作用调控元件控制。最重要的是,组蛋白修饰和/或 DNA 甲基化(histone modifications and/or DNA methylation)等过程对转录具有至关重要的调节影响。如果转录机器无法访问某个区域,例如如果染色质结构因特定组蛋白修饰的存在而被压缩,或者如果启动子 DNA 被甲基化,则转录可能根本无法开始。最后,基因活性还受到转录后 ncRNA(例如 microRNA (miRNA))以及细胞信号传导的控制,从而导致蛋白质修饰或蛋白质-蛋白质相互作用的改变。
1.2.1.1 转录因子通过调控区进行调控
转录因子是识别特定 DNA 基序并与调控区结合并调控与该调控区相关基因转录速率的蛋白质(参见图 1.5 的说明)。这些因子与图 1.5 中总结的各种调控区域结合,它们的协同作用控制着转录速率。除了它们的结合偏好之外,它们的浓度以及协同或竞争转录因子的可用性也会影响转录速率。
1.2.1.1.1 核心启动子和近端启动子(Core and proximal promoters)
核心启动子是转录起始位点 (TSS) 周围紧邻的区域,充当转录机制和起始前复合物 (PIC) 组装的对接位点。转录起始的教科书模型如下:核心启动子在起始序列(Inr)上游30 bp处有一个TATA基序(称为TATA-box),其中也包含TSS。首先,转录因子 TFIID 与 TATA-box 结合。接下来,招募通用转录因子并在起始序列上启动转录。除了TATA-box和Inr之外,动物核心启动子上还有许多与转录起始和PIC组装相关的序列元件,例如下游启动子元件(DPE)、BRE元件和CpG岛。在黑腹果蝇的 TATA-less 启动子中,DPE 位于 TSS 下游 28-32 bp 处。它们通常与 Inr 元素同时出现,并且被认为具有与 TATA-box 类似的功能。 BRE 元件被 TFIIB 蛋白识别,位于 TATA-box 的上游。 CpG 岛是脊椎动物基因组中富含 CG 二核苷酸的片段,尽管这些基因组中 CG 二核苷酸普遍缺失。人类基因组中 50% 至 70% 的启动子与 CpG 岛相关。近端启动子元件通常位于核心启动子的正上游,通常包含激活转录因子的结合位点,并提供对基因表达的额外控制。
1.2.1.1.2 增强子 (Enhancers Proximal)
近端调控并不是基因调控的唯一或最重要的模式。人类基因组中的大多数转录因子结合位点都存在于基因间区域或内含子中。这表明远端调控元件在动物基因组中的广泛使用。在分子功能水平上,增强子与近端启动子相似;它们含有相同转录激活剂的结合位点,并且它们基本上增强了基因表达。然而,它们通常是高度模块化的,其中几个可以同时或在不同的时间点或组织影响相同的启动子。此外,它们的活性与它们的方向和与它们相互作用的启动子的距离无关。许多研究表明,增强子可以作用于数千碱基之外的目标基因。根据一个流行的模型,增强子通过使 DNA 成环并与其目标基因接触来实现这一点。
1.2.1.1.3 消音器 (Silencers )
消音器与增强器类似;然而,它们对靶基因转录的作用与增强子相反,并导致其转录水平降低。它们含有抑制性转录因子的结合位点。阻遏转录因子可以阻断激活剂的结合,直接竞争相同的结合位点,或者诱导抑制染色质状态,在这种状态下激活剂不可能结合。沉默子效应与增强子的类似,与目标基因的方向和距离无关。与这种普遍观点相反,果蝇有两种类型的消音器:远程消音器和短程消音器。短程消音器靠近启动子,长程消音器可以使多个启动子或增强子沉默超过千碱基。与增强子一样,与阻遏子结合的沉默子也可能通过循环和创建高阶结构来诱导 DNA 结构的变化。此类阻遏蛋白中的一类是多梳族蛋白 (PcG),它被认为通过循环启动高阶结构。
1.2.1.1.4 绝缘体 (Insulators)
绝缘体区域将其他调控元件的影响限制在某些染色体边界内;换句话说,它们创建了不受该域外区域监管要素污染的监管域。绝缘体可以阻断增强子-启动子之间的通讯和/或防止抑制性染色质结构域的扩散。在脊椎动物和昆虫中,一些经过充分研究的绝缘体被 CTCF(CCTCC 结合因子)结合。来自不同哺乳动物组织的全基因组研究证实,CTCF 结合在很大程度上是细胞类型不变的,并且 CTCF 基序位置在脊椎动物中是保守的。目前解释绝缘体功能的模型有两种;最流行的模型声称绝缘体通过修改染色体结构来创建物理上独立的域。这被认为是通过 CTCF 驱动的染色质环来实现的,最近的证据表明 CTCF 可以通过创建染色质环来诱导更高阶的染色体结构。根据第二种模型,绝缘体结合的激活剂不能结合增强子;因此,实现了增强子阻断活性,并且绝缘体还可以招募活性组蛋白结构域,为增强子发挥作用创造一个活性结构域。
1.2.1.1.5 基因座控制区 (Locus control regions)
基因座控制区(LCR)是控制基因座上整套基因的不同调控元件的簇。 LCR 帮助基因实现其时间和/或组织特异性表达程序。 LCR可能由多个顺式调控元件组成,例如绝缘子和增强子,它们甚至可以在远距离作用于目标。然而,LCR 以方向依赖的方式发挥作用,例如,如果倒置,β-珠蛋白 LCR 的活性就会丧失。 LCR 功能的机制在其他方面似乎与上述其他远程调节器相似。越来越多的证据表明,DNA 环形成了一种染色体结构,其中靶基因聚集在一起,这似乎对于维持开放的染色质结构域至关重要。
1.2.1.2 表观遗传调控 (Epigenetic regulation)
生物学中的表观遗传学通常是指影响基因调控的DNA序列以外的结构(染色质结构、DNA甲基化等)。本质上,表观遗传调控是DNA包装和结构的调控,其结果是基因表达调控。一个典型的例子是,细胞核内的 DNA 堆积可以通过为转录因子结合创造可接近的区域来直接影响基因表达。表观遗传调控有两种主要机制:i) DNA 修饰和 ii) 组蛋白修饰。
1.2.2 转录后调控
1.2.2.1 非编码RNA的调控
近年来,非编码RNA(ncRNA)相关研究呈爆炸式增长。许多出版物将 ncRNA 视为重要的调控元件。植物和动物产生许多不同类型的 ncRNA,例如长非编码 RNA (lncRNA)、小干扰 RNA (siRNA)、微小 RNA (miRNA)、启动子相关 RNA (PAR) 和小核仁 RNA (snoRNA)(Morris 和 Mattick,2014) )。通过与染色质重塑因子相互作用进行遗传调控,它们在基因调控中发挥作用。 siRNA是短双链RNA,参与基因调控和转座子控制;它们通过与 Argonaute 蛋白合作来沉默目标基因。 miRNA 是短单链 RNA 分子,通过使用其互补序列与其靶基因相互作用,并标记它们以便更快降解。 PAR 也可以调节基因表达:它们是大约 18 至 -200 bp 长的 ncRNA,源自编码基因的启动子(Morris 和 Mattick,2014)。 snoRNA 也被证明在基因调控中发挥作用,尽管它们大多被认为引导核糖体 RNA 修饰(Morris 和 Mattick,2014)。
1.2.2.2 剪接调控 (Splicing regulation)
剪接是由mRNA前体上的调控元件和与这些元件结合的蛋白质来调控的。调节元件分为剪接增强子和阻遏子。它们可以位于外显子或内含子中。根据其活性和位置,有四种类型的剪接调节元件:
• 外显子剪接增强子(ESE)
• 外显子剪接沉默子(ESS)
• 内含子剪接增强子(ISE)
• 内含子剪接沉默子(ISS)。
大多数剪接阻遏蛋白是异质核核糖核蛋白(hnRNP)。如果剪接阻遏蛋白结合沉默元件,它们会减少附近位点被用作剪接点的机会。相反,剪接增强子是剪接激活蛋白结合的位点,并且在该区域上的结合增加了附近位点被用作剪接连接点的可能性(Wang 和 Burge,2008)。大多数与剪接增强子结合的激活蛋白都是 SR 蛋白家族的成员。此类蛋白质可以识别特定的RNA识别基序。通过调节剪接,可以跳过或包含外显子,从而创造蛋白质多样性(Wang 和 Burge,2008)。
1.3 塑造基因组:DNA 突变 (Shaping the genome: DNA mutation)
人类和黑猩猩的基因组相似度高达 98.8%。 1.2% 的差异是我们与黑猩猩的区别。就进化距离而言,与人类物种的距离越远,差异就越大。然而,即使在同一物种的成员之间,基因组序列也存在差异。这些差异是由于一种称为突变的过程造成的,该过程驱动了个体之间的差异,但也为作为遗传变异来源的进化提供了燃料。具有有益突变的个体可以比其他人更好地适应周围环境,并且随着时间的推移,这些有利于生存的突变会由于“自然选择”的过程而在人群中传播。选择作用于具有有益特征的个体,这使它们在特定环境中具有生存优势。个体突变产生的遗传变异提供了选择起作用的材料。如果选择过程在需要适应的相对孤立的环境中持续很长时间,那么只要有足够的时间,这个种群就可以进化成不同的物种。简而言之,这就是进化背后的基本思想,如果没有突变提供遗传变异,就不会有进化。基因组突变的发生有多种原因。首先,DNA复制并不是一个没有错误的过程。在细胞分裂之前,DNA 的复制每 10^8 到 10^10 个碱基对有 1 个错误。其次,紫外线等诱变剂可以诱导基因组突变。导致突变的第三个因素是不完善的 DNA 修复。每天,任何人体细胞都会遭受多次 DNA 损伤。 DNA修复酶的存在是为了应对这种损伤,但它们也不是没有错误的,根据使用哪种DNA修复机制(有多种),错误的发生率会有所不同。
突变根据其影响的碱基数量、对 DNA 结构和基因功能的影响进行分类。根据突变对 DNA 结构的影响,突变可分为以下几类:
• 碱基替换:一个碱基被另一个碱基改变。
• 删除:删除一个或多个碱基。
• 插入:新碱基插入基因组中。
• 微卫星突变:小的串联重复DNA 片段的小插入或缺失。
• 反转:DNA 片段将其方向改变180 度。
• 易位:DNA 片段移动到基因组中的另一个位置。突变还可以根据其大小进行如下分类: • 点突变:涉及一个碱基的突变。取代、删除和插入都是点突变。它们也被称为单核苷酸多态性(SNP)。
• 小规模突变:涉及多个碱基的突变。
• 大规模突变:涉及更大染色体区域的突变。转座元件插入(基因组的一个片段跳转到基因组中的另一个区域)和片段复制(一个大区域串联复制多次)是典型的大规模突变。
• 非整倍体:整个染色体的插入或删除。
• 全基因组多倍体:涉及全基因组的重复。突变可以根据其对基因功能的影响进行如下分类:
• 功能获得突变:一种突变,其中改变的基因产物具有新的分子功能或新的基因表达模式。
• 功能丧失突变:导致蛋白质功能降低或消失的突变。这是更常见的突变类型。
1.4 基因组学中的高通量实验方法(High-throughput experimental methods in genomics )
上述大多数与转录、基因调控或DNA突变有关的生物现象都可以使用高通量实验技术在整个基因组上进行测量,这正迅速成为研究基因组生物学的标准。此外,它们在临床上的应用也正在蓬勃发展,因为已经有基于这些技术的诊断测试。可以通过高通量检测测量的一些内容如下:
• 哪些基因被表达以及表达量如何?
• 转录因子在哪里结合?
• 基因组中哪些碱基被甲基化?
• 翻译了哪些文字记录?
• RNA 结合蛋白在哪里结合?
• 表达哪些microRNA?
• 基因组的哪些部分相互接触?
• 基因组中的突变位于何处?
• 基因组的哪些部分没有核小体?
使用现代全基因组技术可以回答更多问题,并且每隔一天就会出现现有技术的新变体来回答新问题。然而,必须记住,这些方法的成熟程度各不相同,并且都存在技术限制,并且并非没有噪音。尽管如此,它们对于研究和临床目的非常有用。而且,借助这些方法,我们能够大规模地对基因组进行测序和注释。
1.4.1 高通量技术背后的总体思路
高通量方法旨在量化或定位包含感兴趣的生物学特征(表达基因、结合位点等)的全部或大部分基因组。大多数方法依赖于对目标生物学特征的某种富集。例如,如果您想要测量蛋白质编码基因的表达,您需要能够提取具有蛋白质编码基因获得的特殊转录后改变的 mRNA 分子,正如许多 RNA 测序 (RNA-seq) 实验中所做的那样。如果您正在寻找转录因子结合,则需要富集目标蛋白质结合的 DNA 片段,就像 ChIP-seq 实验中所做的那样。这部分取决于现有的分子生物学和化学技术,这部分的最终产物是RNA或DNA片段。
接下来,您需要能够判断这些片段来自基因组中的何处以及有多少。在测序技术普及之前,微阵列(Microarrays)一直是定量步骤的标准工具。在微阵列中,人们必须设计互补碱基,称为“oligos”或“probes”,以通过实验方案富集遗传物质。如果富集的物质与“oligos”互补,则会产生光信号,并且信号的强度将与与该“oligos”配对的遗传物质的量成正比。将有更多的探针(probes)可用于杂交(互补碱基形成键的过程),因此可用的片段越多,信号越强。为了使其发挥作用,您需要至少了解部分基因组序列并设计探针。如果您想测量基因表达,您的探针应该与基因重叠,并且应该足够独特,不会与其他基因的序列结合。该技术现已被测序技术所取代,您可以直接对遗传物质进行测序。如果您有片段的序列,您可以将它们与基因组对齐,查看它们来自哪里,并对它们进行计数。这是一种更好的技术,其中定量基于片段的真实身份,而不是基于与设计探针的杂交。总而言之,HT 技术有以下步骤,图 1.6 也总结了这一点:
• 提取:这是提取感兴趣的遗传物质(RNA 或DNA)的步骤。
• 富集:在此步骤中,您可以富集您感兴趣的事件。例如,蛋白质结合位点。在某些情况下,例如全基因组 DNA 测序,不需要富集步骤。您只需获取基因组 DNA 片段并对它们进行测序即可。
• 量化:您可以在此处量化您的浓缩材料。根据实验方案,您可能还需要量化控制集,在该控制集中您应该看不到富集或仅看到背景富集。
1.4.2 高通量测序 (High-throughput sequencing)
高通量测序,或大规模并行测序,是可以一次对数千/数百万个 DNA 片段进行测序的方法和技术的集合。这与一次只能产生有限数量碎片的旧技术形成鲜明对比。这里,吞吐量是指每小时测序的碱基数。与现代高通量方法相比,较旧的低通量测序方法的通量低约 100 倍。吞吐量的增加使得能够在更短的时间内测量全基因组范围内的生物特征。
与其他高通量方法类似,基于测序的方法也需要富集步骤。此步骤丰富了我们感兴趣的特征。基于测序的方法的主要区别在于量化步骤。在高通量测序中,富集的片段通过测序仪,测序仪输出片段的序列。由于当前领先技术的限制,只能从输入片段中对有限数量的碱基进行测序。然而,长度通常足以将读数唯一地映射到基因组并量化输入片段。
1.4.2.1 高通量测序数据
如果有可用的基因组,则将读数与基因组进行比对,并根据文库制备方案,应用不同的策略进行分析。测序文库由可供测序的 RNA 或 DNA 片段组成。文库的准备主要取决于感兴趣的实验。有许多文库制备方案旨在量化来自基因组的不同信号。图 1.7 描述了不同文库制备方案和读比对处理输出的一些潜在分析策略。例如,我们可能对量化基因表达感兴趣。该实验方案称为 RNA 测序 (RNA-seq),可富集来自蛋白质编码基因的 RNA 片段。比对后,我们可以计算覆盖率概况,从而获得基因组中每个碱基的读取计数。该信息可以存储在文本文件或专用文件格式中,以便在后续分析或可视化中使用。我们还可以计算每个基因的外显子有多少个读数重叠,并记录每个基因的读数计数以供进一步分析。这本质上会生成一个包含不同样本的基因名称和读数计数的表。
正如我们将在后面的章节中看到的,这是统计模型的基本信息用于RNA-seq数据。此外,我们可以堆叠读数并计算读数中的碱基位置与基因组中的碱基不匹配的次数。读取对齐器允许不匹配,因此我们可以看到不匹配的读取。该信息可用于识别SNP,并可再次以表格格式存储,其中包含位置和错配类型以及支持错配的读数的信息。原始算法比仅计算不匹配的算法要复杂一些,但总体思路是相同的;他们所做的不同之处是尝试通过使用过滤器来最大程度地减少误报率,这样就不会将每个不匹配都记录为 SNP。
1.4.2.2 高通量测序的未来
测序技术仍在不断发展。获得更长的单分子读数,并且最好能够动态调用碱基修改是下一个前沿。读长越长,重复含量高的区域的基因组组装就越容易。通过单分子测序,我们将能够判断给定细胞群中存在多少转录本,而无需依赖可能引入偏差的片段扩增方法。最近的另一个发展是单细胞测序。目前的技术通常适用于数千至数百万个细胞的遗传物质。这意味着您收到的结果代表了实验中使用的细胞群。然而,同一类型的细胞之间存在很大的变异,但这种变异根本观察不到。较新的测序技术可以作用于单个细胞并提供每个细胞的定量信息。
1.5 基因组学的可视化和数据存储库
截至 2016 年,大约有 100 个动物基因组被测序。除此之外,还有来自各个实验室或联盟的许多研究项目,产生 PB 级的辅助基因组学数据,例如 ChIP-seq、RNA-seq等。要能够可视化基因组及其相关数据,有两个要求:1)您需要能够使用具有已测序基因组的物种,2)您希望对该基因组进行注释,这意味着,至少,你想知道基因在哪里。大多数基因组在测序后都会快速注释基因预测或将已知的基因序列映射到它们上,您还可以对其他物种进行保护以过滤功能元素。如果您正在研究模型生物体或人类,您还将获得大量辅助信息来帮助划分功能区域,例如人群中常见的调节区域、ncRNA 和 SNP。或者您可能有可用的疾病或组织特异性数据。对有机体的研究越多,您获得的辅助数据就越多。
1.5.0.1 通过基因组浏览器访问基因组序列和注释
UCSC 基因组浏览器:这是由加州大学圣克鲁斯分校托管的在线浏览器,网址为 http://genome.ucsc.edu/。这是一个交互式网站,包含许多物种的基因组和注释。您可以搜索您感兴趣的物种的基因或基因组坐标。它通常响应速度非常快,并且允许您可视化大量数据。此外,它还有多个其他工具可以与浏览器结合使用。最有用的工具之一是 UCSC 表浏览器,它允许您以多种格式下载在浏览器上看到的所有数据,包括序列数据。用户可以上传数据或提供数据链接以可视化用户特定的数据。
Ensembl:这是欧洲生物信息学研究所和英国 Wellcome Trust Sanger 研究所维护的另一个在线浏览器,http://www.ensembl.org。与 UCSC 浏览器类似,用户可以可视化多个物种的基因或基因组坐标,并且还附带辅助数据。 Ensembl 与 Biomart 工具关联,该工具类似于 UCSC Table browser,可以下载多种格式的基因组数据,包括所有辅助数据集。
IGV:综合基因组查看器 (IGV) 是由 Broad 研究所 (https://www.broadinstitute.org/igv/) 开发的桌面应用程序。它是为处理大量高通量测序数据而开发的,这些数据很难在在线浏览器中查看。 IGV 可以将您的本地测序结果与台式机上的在线注释集成。这在查看测序数据(尤其是比对)时非常有用。上面提到的其他浏览器也有类似的功能,但是您需要先在某个地方在线提供大型测序数据,然后才能通过浏览器查看。
1.5.0.2 高通量检测的数据存储库
基因组浏览器包含大量辅助高通量数据。然而,还有更多可用的公共高通量数据集,并且它们肯定无法通过基因组浏览器获得。通常,与出版物相关的每个高通量数据集都应存放在公共档案中。我们使用两个主要的公共档案馆来存放数据。其中之一是托管在 http://www.ncbi.nlm.nih.gov/geo/ 的基因表达综合 (GEO),另一个是托管在 http://www.ebi 的欧洲核苷酸档案 (ENA)。 ac.uk/ena。这些存储库接受高通量数据集,用户可以免费下载和使用这些公共数据集进行自己的研究。这些存储库中的许多数据集都是原始格式,例如,排序器主要提供的格式。一些数据集也会有经过处理的数据,但这不是常态。
除了这些存储库之外,还有多个致力于某些基因组生物学或疾病相关问题的跨国联盟,它们维护自己的数据库并提供对已处理数据和原始数据的访问。下面提到了其中一些联盟。