从零开始的“RNA-seq“分析(STAR进行mapping)
前言
在用trim_galore进行trimming后,要进行RNA-seq分析的核心部分:mapping
实验室一直购买着CLC Genomics Workbench的版权,用户友好,使用简便,但一年大概要花费30万日币左右,这次挑战用完全免费的Linux+STAR来建立index进行mapping,减少实验开支,给我打钱就可以。
软件的下载和安装
2021/12/7 当前最新版本是STAR-2.7.9
下载安装的方法主要参考STAR的官方网站就可以了。https://github.com/alexdobin/STAR
# Get latest STAR source from releases
wget https://github.com/alexdobin/STAR/archive/2.7.9a.tar.gz
tar -xzf 2.7.9a.tar.gz
cd STAR-2.7.9a# Compile under Linux
cd /STAR-2.7.9a/source
make STAR
#test
STAR如果出现
Usage: STAR [options]... --genomeDir /path/to/genome/index/ --readFilesIn R1.fq R2.fq
Spliced Transcripts Alignment to a Reference (c) Alexander Dobin, 2009-2019
For more details see:
To list all parameters, run STAR --help
那就说明安装成功了。
建立index
建立index的第一步是从网站上下载注释及genome文件,因为我基本上只做老鼠和人的RNA-seq,使用了权威的Genecode数据库:GENCODE - Home page
这次以老鼠为例:
下载注释文件(annotation)
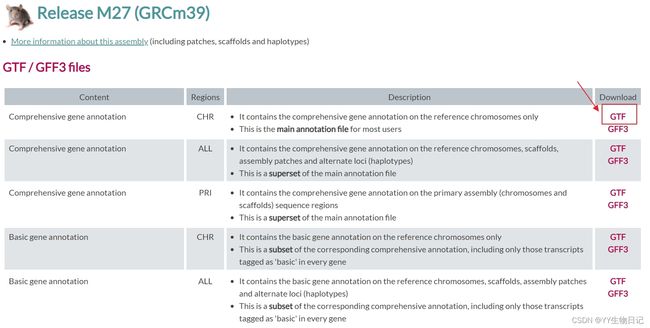
下载fasta基因文件

然后运行STAR的genomeGenerate来建立index
事先创建一个index的文件夹(下面代码是建立人的基因组index时使用的!)
STAR --runThreadN 10
--runMode genomeGenerate
--genomeDir index/
--genomeFastaFiles GRCh38.p13.genome.fa
--sjdbGTFfile gencode.v38.annotation.gtf
--sjdbOverhang 35
–runThreadN :线程数 (不清楚的自己查看一下电脑的CPU信息)
–genomeDir :index输出的路径 (事先准备好的index文件夹)
–genomeFastaFiles :参考基因组(之前下载的.fa文件)
–sjdbGTFfile :参考基因组注释文件 (之前下载的.gtf文件)
–sjdbOverhang :readlength -1 (默认值是100)
sjdbOverhang 这次设定为35,因为这次的read length都为36bp
进行Mapping
STAR --runThreadN 10
--runMode alignReads
--readFilesCommand zcat
--twopassMode Basic
--outSAMtype BAM SortedByCoordinate
--genomeDir /reference/genome/grcm39/index/
--readFilesIn 2_R1_val_1.fq.gz 2_R2_val_2.fq.gz
--outFileNamePrefix /wkdir/WT2Brain--runThreadN: 使用线程数
--readFilesCommand: 这次的read file是fq.gz文件,这里要用zcat
--twopassMode Basic: 这次使用twopassmode来mapping
--genomeDir: 之前建立的index文件夹路径
--readFilesIn: 跑mapping的sample路径
--outFileNamePrefix: 输出文件的路径,我个人喜欢分文件夹来保存不同sample的mapping结果,一起在一个有一点乱。
--outSAMtype:BAM SortedByCoordinate,输出排序的BAM file,BAM可以理解为SAM的二进制格式,占用空间小,按照基因组排不排序不知道对后面解析有什么影响,但数据不会失真。
还有好多可设置的parameter,有时间还是把STAR的manual仔细阅读为好。
Dec 07 18:03:31 ..... started STAR run
Dec 07 18:03:31 ..... loading genome
Dec 07 18:05:35 ..... started 1st pass mapping
Dec 07 18:07:22 ..... finished 1st pass mapping
Dec 07 18:07:22 ..... inserting junctions into the genome indices
Dec 07 18:08:19 ..... started mapping
Dec 07 18:10:16 ..... finished mapping
Dec 07 18:10:18 ..... started sorting BAM
Dec 07 18:11:13 ..... finished successfully
有点意外,只调用了10个线程,一个sample 2-pass-mode居然8分钟就跑完了。
接下来准备用Qualimap 来做质控分析。