iMeta | 青岛华大范广益组基于共标签测序数据的高质量宏基因组组装工具MetaTrass...
点击蓝字 关注我们
MetaTrass:基于共标签测序数据的人类肠道微生物高质量宏基因组组装工具

https://doi.org/10.1002/imt2.46
RESEARCH ARTICLE
●2022年8月15日,青岛华大基因研究院齐彦伟团队在iMeta在线发表了题为“MetaTrass: a high-quality metagenome assembler of the human gut microbiome by cobarcoding sequencing reads”的文章。
● 该研究发表了宏基因组组装工具MetaTrass,采用先读长分类分箱后组装策略,通过综合利用共标签测序数据和参考基因组信息实现高效率地组装人类肠道微生物群落菌种水平的高质量基因组。
● 第一作者:齐彦伟、古盛强
● 通讯作者:范广益([email protected])、邓礼([email protected])
● 其他作者:张跃、郭立东、徐梦阳、程小芳、王欧、孙颖、陈建威、方晓东、刘心
亮 点
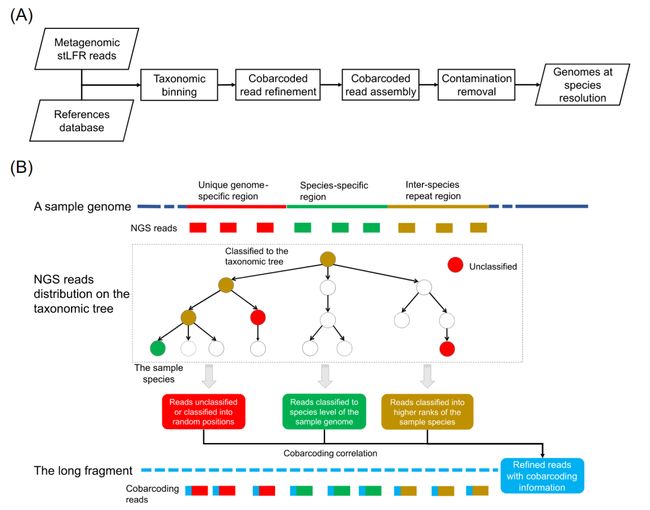
● MetaTrass采用先读长分类分箱后组装策略的宏基因组组装工具,其源代码已在Github网址https://github.com/BGI-Qingdao/MetaTrass 公开
● MetaTrass通过综合利用共标签测序数据和参考基因组信息实现高效率地组装人类肠道微生物群落菌种水平的高质量基因组
● MetaTrass利用二代测序短读长之间的共标签关联来减少传统分箱结果中的假阴性率,并改善菌种草图的组装连续性
摘 要
大量基于宏基因组学的研究展示了人类肠道微生物基因组非保守区域内的巨大遗传多样性,所以亟需高效的方法来获取微生物的高质量基因组以在菌种级的分辨率下阐明微生物群落中的变异性规律。然而,从宏基因组测序数据集中获取数目足够的高质量微生物基因组仍然是生物信息学分析中的巨大挑战。在此,我们开发了名为MetaTrass的宏基因组组装工具。该工具综合利用单管长片段测序数据和公开的微生物参考基因组信息,并采用先测序读长分类分箱再组装的策略实现菌种级的高质量基因组的组装。单管长片段测序技术是深圳华大基因研究院自主研发的一类具有超大标签数的共标签测序技术。相对于传统的先组装再分箱策略,MetaTrass能够从宏基因组测序数据中组装出更多长连续性、高完整度、低污染度的微生物基因组。在一个人工模拟微生物菌群的测试中,相对于基于传统的二代测序数据的组装方法,MetaTrass能够将菌种基因组的组装连续性从kb提升到Mb,同时保持相近的准确度。在4个真实人类肠道微生物样本数据集的测试中,MetaTrass成功组装出178个高质量基因组,而在传统策略下每个样本的最优结果之和仅有58个。重要的是,通过该结果我们能确认不同样本对应的基因组层级的微生物组成结构以及不同样本共有菌种之间的基因差异等。MetaTrass不仅适用于单管长片段测序数据同时也适用于其它类型的共标签测序数据。由于它具有超强的组装高质量宏基因组的能力,MetaTrass将有力地促进微生物群落的不同时间、空间动态特征的高精度基因组学研究。MetaTrass的开源代码可以在Github网址https://github.com/BGI-Qingdao/MetaTrass免费获取。
视频解读
Bilibili:https://www.bilibili.com/video/BV1NU4y1y7Sj/
Youtube:https://youtu.be/R1L9fw_uggE
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
宏基因组学通过直接对环境中非分离培养的微生物群落进行DNA测序以及分析的方法,其研究结果推进了人们对于非分离培养微生物的认识和研究。随着综合性宏基因组数据的增加,微生物菌落,特别是人肠道微生物的基因多样性被广泛报道。宏基因组学相关的进展为研究人类肠道复杂微生物群落的空间分布和动态提供了新的可能性。
通过对高质量菌株水平的基因组进行功能挖掘,研究人员发现菌株之间的基因型差异与它们的表型差异有很强的关联性。种内非同源基因可以作为生物标志物来区分一个菌种内的致病菌株和其相应的共生菌株。研究表明菌种间共享的保守种内同源序列的比例可能低至40%,同时其余的非保守基因组序列认为是微生物的表型多样性重要根源。因此,来自微生物样本的菌种级完整基因组将使人们能够更全面地认识菌种内的遗传多样性。但是从宏基因组测序数据集中获取足够的高质量基微生物因组仍然是生物信息学分析的巨大挑战。
目前大多数分析微生物群落的方法都是基于低成本、高通量的第二代测序技术(NGS)设计的。研究人员开发了大量高度模块化的具有不同功能的计算工具,包括基因组组装工具、基因组分箱工具、测序数据分箱工具和分类轮廓分析工具。其中,先组装后基因组分箱的策略已被普遍用于宏基因组组装中,从宏基因组测序数据中组装获取的菌种基因组被称为宏基因数据组装基因组(MAG)。在传统的策略中,首先由宏基因组组装工具在考虑不同菌种具有不均匀测序覆盖深度的条件下将来自微生物群落的短读长组装成长序列,并进一步由基因组分箱工具根据K-mer组成和读长覆盖率等将组装结果分箱成不同的单个菌种的基因组。然而,这些传统策略在构建重叠群时往往不能有效地解决由种间的长重复序列带来的困难。因此,从NGS的宏基因组测序数据中组装得到的基因组草图的连续性仍然不够长,导致其难以用于研究微生物基因组的大尺度结构变异。
为了克服长重复序列导致的问题,研究人员开发出各种具有长距离信息的测序技术以及相应的计算工具。如:太平洋生物科学公司和牛津纳米孔技术公司(ONT)开发的第三代单分子实时测序(TGS)技术可以产生长度达数百kbp的连续读长。在组装高连续性的完整基因组方面,不同的研究显示了它们对培养和非培养的微生物都具有应用巨大潜力。利用高通量染色体构象捕获(Hi-C)技术产生的染色质结构信息,研究人员也可以组装出更多具有更高连续性的高质量微生物宏基因组。对于NGS数据集,多个样本中相似菌种间的共同丰度也被用来提高的宏基因组组装的质量。然而,以上的方法都存在一定的局限性。TGS长读数中的高测序错误率增加了区分真正的生物变异和测序错误的难度。Hi-C数据的有效利用则以利于长连续性的基因组草图。利用多个样本中菌种的丰度关联的方案则会忽略单个样本的基因组特征,同时增加相应的测序成本。
共标签测序文库是基于短读长测序文库改进的新型测序技术,其包含的共标签信息是一类非连续的基因组长程信息,可以为改进宏基因组分析提供不同的途径。在共标签文库的构建中, DNA分子的长片段首先被分配到具有不同化学反应环境的物理空间,并在不同的物理空间进一步剪切成较短的子片段。然后这些子片段将被插入相同的标签序列。最后,通过标准的短读长测序平台对具有标签序列的子片段进行测序。对于不同的共标签文库技术,如BGI的单管长片段读长技术(stLFR)、10X Genomics的链接读长技术和Illumina的连续性保留转座酶测序技术,标签序列的数量和长片段的短读长覆盖率等技术指标对下游分析有很大影响 。共标签技术的共标签信息在基因组草图序列或组装图上的统计关联已被应用于大型真核生物基因组和宏基因组的组装中,并能显著地提升组装连续性。然而,这些策略都难以有效地解决微生物群落中丰度不均的菌种间长重复序列带来的组装难题。
在这项工作中,我们基于共标签测序数据和参考基因组开发了名为宏基因组单菌种水平分类读长组装工具,简称为MetaTrass。与传统的策略不同,MetaTrass的特点是采用先测序读长分类分箱后基因组组装的综合性方法。共标签信息在该策略中主要有两个作用。首先,它通过对具有相同标签序列进行聚类以改善了最初的测序读长分类分箱效果。其次,它利用共标签的长程信息提升组装的连续性。为了评估它的产生具有长连续性和高分辨率的高质量基因组草案的效率,我们在一个人工模拟微生物群落和四个人类肠道微生物群落的stLFR数据集上测试了MetaTrass。基于MetaTrass组装的高质量基因组草图,我们定量地分析了四个人类肠道生物群落的微生物组成和遗传多样性。由于我们先执行了测序读长分类分箱过程,所以MetaTrass组装的高质量基因组草图具有明确的菌种分箱信息,这也将有利于微生物的下游分析。同时,我们将MetaTrass与现有的主流工具进行了综合性比较来确定它的效率。
方 法
数据集
在该工作中,我们利用一个人工模拟微生物菌群和四个人类肠道微生物菌群数据来评估MetaTrass的效率。人工模拟微生物样本(ZymoBIOMICSTM Microbial Community DNA Standard,目录D6305,批号ZRC190812)包括8个细菌和2个真菌,它们的平均丰度分别约为12%和2%(表S4)。四个人类肠道微生物菌群是从三个健康志愿者和一个炎症性肠病患者的粪便中提取的。根据stLFR技术的标准实验方案,我们构建了所有样本的stLFR文库。首先,DNA样本被随机剪切成长片段,然后长片段被带有独特标签序列的磁珠捕获。最后,每个长片段在磁珠的微环境下被微珠表面的Tn5转座酶进一步分割并插入相同的标签序列。人工模拟微生物样本和患者样本的stLFR文库在BGISEQ-500平台上进行测序,而健康人样本的stLFR文库在MGISEQ-2000平台上进行测序。所有数据集的双端测序读长的长度为100bp。三个健康人样本的文库被分别分配到半个泳道,每个样本产生约50Gb的原始数据。人工模拟样本的文库和病人样本的文库分别被分配到一个完整的泳道上,分别产生约85和100Gb的原始数据。stLFR原始数据中,标签序列位于从双端测序数据中反向读长的末端。我们用已公开的工具stLFR_barcode_split对原始stLFR数据进行拆分,将标签序列替换成数字符号并添加到FASTQ文件中的读长名中。SOAPfilter_v2.2被用来去除带有引物序列、低质量碱基过多和高重复率的低质量原始读长,其参数设置为(-y -F CTGTCTCTTATACACATCTTAGGAAGACAAGCACTGACGACATGA -R TCTGCTGAGTCGAGAACGTCTCTGTGAGCCAAGGAGTTGCTCTGG -p -M 2 -f -1 -Q 10)。最后,人工模拟微生物样本保留了59.45Gb的干净数据,第一个健康人粪便样本(H_Gut_Meta01)保留了34.48Gb,第二个健康人粪便样本(H_Gut_Meta02)保留了35.33Gb,第三个健康人粪便样本(H_Gut_Meta03)保留了37.88Gb,而病人样本(P_Gut_Meta01)保留了97.20Gb。所有样本的干净数据集都可以在中国国家基因库(CNGB)的序列归档系统(CNSA)(https://db.cngb.org/cnsa/)中找到,其对应的登记号为CNP0002163。
读长分类分箱
我们采用Kraken2(2.0.9-beta版)将stLFR读长分箱到不同的菌种。首先,我们需要针对待研究的微生物群落选择相应的参考基因组集合并构建定制化的Kraken数据库。然后,根据数据库对stLFR读长进行分箱。在这两步,Kraken都以默认参数运行。具体来说,ZYMO产品所附的参考基因组被用来构建人工模拟样本的Kraken数据库。而人肠道微生物的Kraken数据库则是下载了UHGG数据库。该数据库包含了人肠道菌群中4542个菌种水平的代表性基因组。
共标签读长精炼
Kraken2基于参考基因组的K-mer的共享情况构建了K-mer的分类树,从而减少种间重复序列对应K-mer的多重命中次数。因此,来自重复区域的读长将被分箱到比种更高的最低共同祖先(LCA)等级。有一部分研究也尝试基于测序数据的覆盖深度或共标签信息的统计推断来实现这类读长的分箱。MetaTrass利用被分箱到某一菌种的读长与被归入到更高LCA等级的读长之间的共标签关联来减少被分箱到更高分类等级的假阴性读长。在菌种水平上被分箱到菌种级别的高可信读长被定义为相应菌种的分类读长集(TR)。针对每一个标签,我们根据被归类为TR的读长数(Num_T)和这些读长与总读长数的比率(Ratio_T)来收集和完善每个菌种的读长集合。首先,TR集合中读长所携带的标签被提取出来作为候选标签。然后我们根据Num_T和Ratio_T对标签进行由大到小的排序。最后,那些Num_T和Ratio_T值较高的标签对应的读数优先被收集,最终收集的读长数不超过300×。考虑到足够的读长覆盖深度是组装完整基因组的必要条件,在此步骤中我们仅分析丰度高于10×的菌种。我们通过TRs的总碱基数与参考基因组的碱基数的比值来估计每个菌种的丰度。Seqtk(1.3-r114-dirty版本)被用来提取了每个菌种对应的精炼读长集。需要注意的是,尽管Ratio_T被用来控制假阳性读长的数量,仍然有一些假阳性读长由同一磁珠捕获到多个来自不同菌种的长片段所引入。从这些假阳性读长组装出来的序列将被“去污染”模块进一步过滤掉。
共标签读长组装
在该步骤中,我们用Supernova(2.1.1版)组装丰度高于10×的单个菌种的读长集合,Supernova是能利用共标签信息实现单个大型真核生物基因组从头组装的工具。Supernova是针对10X Genomics (http://www.10xgenomics.com/)的链接读长数据设计的,而链接读长数据的格式与stLFR读长数据集存在一定的差异。因此,在运行Supernova之前,我们需将stLFR读长转换成链接读长的 FASTQ格式。此外,为了让Supernova允许较大的覆盖深度差异,我们将参数--accept-extreme-coverage 设置为 "yes"。
去污染
在stLFR文库中,来自不同菌种的长片段可以被同一个磁珠捕获。虽然该几率很低,但是也会导致来自不同菌种的读长会标记相同的标签序列。因此,经过共标签读长精炼后的读长集的组装草图会包含来自其它菌种的污染序列。我们利用组装草图序列与相应参考基因组的相似性来去除污染序列。在之前的研究中,比对覆盖度(AF)和平均核苷酸一致性(ANI)已被普遍用来界定不同菌种。MetaTrass也采用了AF和ANI的指标。ANI定义为每个比对结果中查询序列与参考序列匹配碱基数与参考序列的碱基数之比,而AF则被定义为大于给定ANI阈值的比对的总长度与序列总长度的比值。在实际的处理过程中,ANI的默认阈值设置为90%,而AF的默认阈值设置为50%。不满足以上阈值的组装序列将被当成污染序列而过滤掉。我们利用QUAST(5.0.2版)获取基因组草图和参考基因组之间的初始比对信息。除了获得有效比对的一致性阈值被设置为90%,其它参数都采用默认值。
先组装后分箱策略
在NGS宏基因组组装的常规分析流程中,研究人员一般先对宏基因组数据进行混合从头组装,然后将组装序列分箱成不同的基因组草图,即先组装后分箱策略。基于一个人工模拟菌群和四个人类肠道微生物菌群的stLFR数据集,我们比较了MetaTrass和由不同组装工具和分箱工具所组合的先组装后分箱的方法。在我们的测试中,不同的stLFR数据集被不同的组装工具所组装。其中NGS组装工具包括IDBA-UD(1.1.3版)、MEGAHIT(1.1.3版)和MetaSPAdes(3.10.1版),而共标签组装工具则包括Supernova、Athena(1.3.0版)和CloudSPAdes(3.13.1版)。然后,我们采用了两个主流的基因组分箱工具对组装结果进行分箱,包括MetaBAT2(2.12.1版)和Maxbin2.0(2.2.5版)。由于Supernova、CloudSPAdes和Athena无法直接使用stLFR读长数据格式,我们利用公开的工具stlfr2supernova_pipeline中的step_2_10X_fake.sh脚本进行了适当的格式转换。除了Supernova之外,所有的组装工具都以默认参数运行。为了使Supernova能够组装具有极高覆盖深度的数据集,参数--accept-extreme-coverage被设置为yes。所有的组装结果都可以在CNGBdb的CNSA(https://db.cngb.org/cnsa/)中找到,登记号为CNP0002163。MetaBAT2和Maxbin2.0也是在默认参数下运行的。
评估
我们利用基于参考基因组和非参考基因组的评估工具评价不同数据集的组装质量。对于人工模拟微生物菌群,我们采用了基于参考基因组的工具QUAST来评估宏基因组组装结果的连续性和准确性。QUAST采用Minimap2(2.17-r974-dirty)将组装序列比对到参考基因组上,并获得碱基一致性大于95%的有效比对。在QUAST的默认参数下,我们评估了基因组覆盖度、NG50/NGA50和组装错误的数量等统计数据。对于人类肠道微生物菌群,我们采用非参考基因组的评估工具CheckM(1.1.2版)来评估每个基因组的完整度和污染度,其运行参数都为默认值。CheckM是以所有细菌中都包含的保守基因作为标记序列进行评估的工具。按照CheckM开发者提出的指导意见,我们定义了高质量的组装(完整度>90%,污染度<5%)和中等质量的组装(完整度>50%,污染度<10%,且不满足高质量条件)。此外,CheckM也可以获得基因组的其它的统计数据,特别是N50、基因组大小和分类等级等,其中分类等级被用来标定组装结果的分辨率。
基因变异以及分类注释
在该工作中,我们对MetaTrass组装的所有高质量基因组都进行了变异分析。我们使用Minimap2将基因组与它们相应的参考基因组进行比对,并设置参数(-x asm5)以防止比对延伸到多样性大于5%的区域。SAMtools(1.9版)和PAFtools(2.17-r982-dirty)被用来将初始未排序的比对结果(BAM格式)转换成按比对位置排序的比对结果(PAF格式)。我们使用PAFtools中的 "call "模块来检测变异,设置参数 “-L 10000”以过滤掉短于10,000 bp的比对。在分析中,SNVs仅包含单核苷酸替换,不包含单碱基的插入或缺失。长度短于50bp的插入或缺失被定义为小变异,其余被定义为大变异。如果某个变异出现在不同样本中的同一菌种,且具有相同位置和序列信息,那么该变异被确定为样本共享变异。
在默认参数条件下,我们使用GTDB-Tk(0.3.1版)的 "classify_wf "功能对MetaTrass和传统策略获得的高质量基因组进行分类注释。基于UHGG数据库的构建原理,如果某个菌种组装结果与近缘菌种的参考基因组的AF高于30%,ANI高于95%,那么该组装结果将分配到菌种水平。根据以上注释信息,我们利用Interactive Tree of Life工具(iTOL 4.4.2版)来可视化高质量组装结果的分类树。
结 果
MetaTrass流程概述
在该工作中,我们基于参考基因组和DNA共标签测序数据的信息开发了名为MetaTrass的宏基因组组装工具。如流程图(图1A)所示,stLFR读长的分类分箱是先于基因组组装的,这与传统的先组装后分箱策略明显不同。在读长的分类分箱中,我们利用Kraken2将宏基因组的stLFR测序数据分箱到不同的分类等级。基于Kraken2的算法原理,只有来自保守的菌种特异区域的读长才能成功地被分箱到相应的菌种。而来自菌种间重复和样本基因组特异区域的读长不能被有效分箱(图1B)。在本文中,菌种特异区域代表了同一物种的不同基因组之间的相似或重复序列。菌种间重复区代表不同菌种的基因组之间的相似或重复序列。独特的基因组特异区域代表了同一菌种的不同基因组之间的差异序列。在Kraken的分箱中,来自菌种间重复区域的读长被分箱到目标菌种的较高分类等级,而来自基因组特异区域的读长将无法被有效分箱。在四个人类肠道微生物样本数据集中,总共有大约10%的读长被归入高于菌种水平的等级,大约9%的读长没有被分箱(表S1)。在共标签读长的精炼步骤中,来自菌种特异区域的读长和来自菌种间重复或样本基因组特异区域的读长之间的共标签关联被用来富集目标菌种的有效读长集(图1B)。首先,我们提取菌种特异读长的标签作为候选标签。然后,在数据大小和共标签信息质量的限制条件下收集最终的标签。其中共标签信息的质量由高可信的菌种分类读长数量以及其在该标签下的占比来量化。最后根据最终的标签收集目标菌种的读长。该步骤所获得的读长集合将在接下来的步骤中进行组装。为减少具有极高丰度菌种所造成计算消耗,我们设置了数据大小阈值来控制单个菌种组装的数据总量。根据样本P_Gut_Meta01中极高丰度菌种在不同深度的组装测试结果(表S2),MetaTrass设置的默认数据大小阈值为300×。在共标签组装中,我们利用Supernova独立组装每个菌种精炼后的读长集合。如前所述,在stLFR文库中来自不同菌种的长片段存在一定的几率共享相同的标签序列(图S1)。因此,上一步组装的基因组草图可能包括少量由交叉标签导致的非目标菌种的读长组装而成的污染序列。为消除污染序列的影响,我们在去除污染的步骤中根据序列与参考基因组的相似性来消除这些污染。我们所采用的参数为平均核苷酸一致性(ANI)和比对覆盖度(AF)。根据样本P_Meta_Gut01(表S3)的测试最佳结果, ANI和AF的默认阈值被设置为90%和50%。总的来说,我们的方法通过综合使用共标签和参考基因组信息既减少了读长分类分箱的假阴性率,同时也降低了共标签读长精炼所带来的假阳性率。
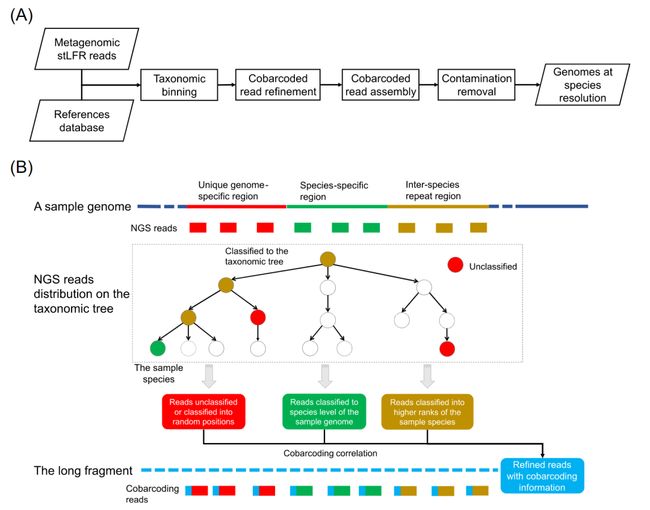
图1. MetaTrass的流程图和原理示意图
(A) MetaTrass组装的流程图。(B) 共标签关联和不同区域读长的分类树分布示意图。
人工模拟微生物数据的组装
先读长分类分箱后组装的策略常被用于组装具有较大基因组的真核生物的单倍型基因组。然而,该策略没有被用于宏基因组组装。为了测试该策略在微生物基因组组装中的效率,我们首先用人工模拟微生物群落的stLFR读长数据集来测试MetaTrass。因为人工模拟微生物群落的简单性,即低种间重复和低基因组特异性区域,总共有高达99.4%的读长被高可信地分箱到菌种水平(表S4)。此外,我们也将MetaTrass与主流的混合组装方法进行了比较(图2A)。除了MetaTrass之外,我们还利用IDBA-UD、MEGAHIT、Supernova、CloudSPAdes和Athena直接组装了该stLFR读长数据集。此外,Nicholls等人的ONT和Illumina NGS读长数据集合的最佳混合组装结果也被用作比较分析,其结果分别由WTDBG和SPAdes进行组装。混合组装方案下,我们利用去污染模块提取了每个菌种的基因组草图。
总的来说,我们的工具在组装具有高完整度和长连续性的基因组草图方面表现优于其它方案(图2)。在Supernova混合组装下,粪肠球菌和发酵乳杆菌的基因组并不完整,它们的基因组覆盖度低至17.7%和8.9%。相反,同样使用Supernova进行组装的MetaTrass则得到了高完整度的结果。这可能是由于丰度不均所引入的组装复杂性被读长的分类分箱所降低。与WTDBG组装TGS读长的结果相比,MetaTrass组装结果的基因组覆盖度更高,且与NGS或为宏基因组设计的共标签组装工具产生的结果相似。与NGS数据的组装工具相比,共标签和TGS数据的组装工具产生的基因组草图具有更好的连续性,其中MetaTrass的性能最好。MetaTrass产生了7个NG50在2Mb左右的组装草图,并获得了最多的高准确度且高连续性的基因组,即它们的NGA50在2Mb左右。此外,与TGS组装工具相比,MetaTrass产生的组装错误更少(图S2)。stLFR读长的组装结果中每100kb的平均错配数和插入缺失分别为60和10,远小于ONT数据集的组装结果。值得注意的是,MetaTrass对铜绿假单胞菌基因组组装的改进并不大,这可能是该菌种的测序数据所包含的有效共标签信息量不足所导致的(表S5)。其中,有效共标签信息是通过计算每个菌种所包含的有效长片段的数量来评估的。有效的长片段被定义为具有超过5个双端测序读长的簇,并且该读长簇在参考基因组上的比对位置中最远的两个双端读长之间的距离超过10kb。

图2. 不同策略的组装方案和结果评估
(A) 基于不同测序数据和组装策略的组装方法。(B) 由QUAST评估所得的基因组覆盖度、NG50和NGA50
四个人肠道微生物菌群数据的组装
为了评估MetaTrass策略的鲁棒性,我们进一步将MetaTrass应用于四个人类粪便样本以研究人类肠道微生物群落。在Kraken2的读长分类分箱过程中,我们采用了统一人类胃肠道基因组数据库(UHGG V1.0)的综合性参考基因组,并通过不同分类等级的分类读长数来估计微生物群落组成结构(图S3-S6)。三个健康样本具有相似的微生物群落,它们主要的微生物群落构成为韧皮菌A 门。而患者样本微生物群落则以变形菌门为主,而变形菌已被证明与肠道微生物菌群失调导致的肠道疾病密切相关。在H_Gut_Meta01、H_Gut_Meta02、H_Gut_Meta03和P_Gut_Meta01样本中,丰度高于10×的菌种数量分别为113、108、93和158。
基因组覆盖度通常被用来评估基因组草图的完整性。四个样本中不同菌种的基因组覆盖度都比较分散,它们分布在0%到90%之间,而H_Gut_Meta01和H_Gut_Meta02的分布比H_Gut_Meta03和P_Gut_Meta01的更集中(图3A)。不过,超过一半的菌种其基因组覆盖度高于50%。考虑到样本基因组和参考基因组之间的巨大遗传差异,这些结果表明我们的工具能有效获得丰度高于10×的菌种的相对完整的基因组信息。同时,四个样本中菌种的基因组覆盖度和组装长度与参考基因组长度的比值之间的差异也展示了样本与参考基因组之间存在着显著的遗传差异(图S7)。四个样本的组装结果中,基因组N50值分散在几kb到几Mb之间,且H_Gut_Meta02和H_Gut_Meta03样本的中位数高于H_Gut_Meta01和P_Gut_Meta01(图3B)。虽然存在以上的差异,但是四个样本组装结果的N50的第三个四分位数都大于100kb,这表明MetaTrass能生成具有高连续性的基因组草图。值得注意的是,在三个健康人样本中,MetaTrass获得了大量具有超长连续性(N50>1Mb)的基因组草图。这些结果将为研究微生物的大尺度基因变异提供了可能性。
考虑到菌种内存在的遗传差异,我们同时利用非基于参考基因组的评估工具CheckM来评估组装结果的质量。在三个健康人样本中,所组装的基因组的完整度中位数都大于92%,而污染度中位数小于2%(图3C)。患者样本的基因组完整度中位数约为83%,污染度中位数约为7%(图S8)。在四个样本中,MetaTrass组装了大量的高质量和中等质量的基因组(图3D)。在H_Gut_Meta01样本中,MetaTrass组装了52个高质量和37个中等质量的基因组,在H_Gut_Meta02中组装了55个和24个,在H_Gut_Meta03中组装了47个和16个,在P_Gut_Meta01中组装了24个和28个。

图3.四个人类肠道微生物数据的MetaTrass组装基因组的QUAST和CheckM评估结果
(A) 基因组覆盖度。(B) Scaffold N50。(C)完整度和污染度的箱型图。(D) 高质量和中等质量基因组的数量。
与先组装后分箱传统策略的比较
为了进一步评估MetaTrass的效率,我们将其与多种传统的先组装后分箱的方法进行了比较。具体的组合情况见方法部分的详细描述。需要注意的是,目前仍然没有直接利用共标签信息进行基因组分箱的工具。从完整性大于50%的基因组数量来看(表S6),MetaTrass在所有数据集上的表现都优于其它方法。特别是对于P_Gut_Meta01,MetaTrass产生了117个完整度高于50%的基因组草图,远多于Supernova和Maxbin2.0的最佳组合所获得的66个。
通过对每个基因组的完整度、污染度和分类等级的综合分析,我们对MetaTrass和传统策略在获得高质量和中等质量基因组的能力以及分类等级的分辨率方面进行了评估(图4)。对于不同的样本,传统方法产生最佳结果的组合是不同的。对于H_Gut_Meta01、H_Gut_Meta02、H_Gut_Meta03和P_Gut_Meta01来说,MetaSPAdes和Maxbin2.0、Supernova和MetaBAT2、MetaSPAdes和MetaBAT2以及Athena和MetaBAT2的组合分别是最佳的。对于这四个样本,传统策略的最优结果都比不过MetaTrass。以H_Gut_Meta01为例,MetaSPAdes和Maxbin2.0的组合产生了41个高质量和中等质量的基因组,远远少于MetaTrass获得的90个。在该组合所得到的18个高质量基因组中,只有3个基因组的分类等级低于目等级,但在MetaTrass中有15个低于目等级。与只使用NGS读长信息的传统策略相比,MetaTrass通过产生更高的连续性和更精细的分辨率而具有优势。这些结果表明MetaTrass使用共标签信息的方式比传统方法更加有效和准确。
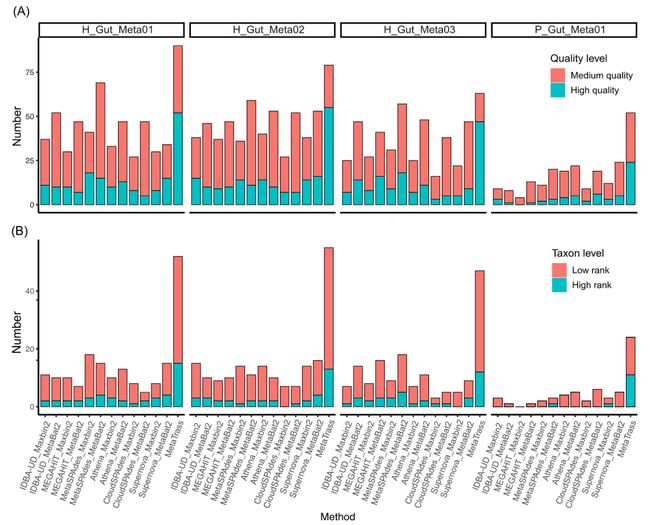
图4. 不同方法组装结果的比较
(A) 不同方法组装得到的高质量和中等质量基因组的数量。(B) 不同方法组装得到的高质量基因组包含的高分类等级和低分类等级的基因组数目。
人类肠道微生物的组成结构包含大量的科学问题而引起了研究人员的关注,其中最有趣的就是它与宿主特征之间存在强烈的关联。为了比较不同方法获得的高质量基因组的微生物组组成,我们统一使用GTDB-Tk对这些基因组进行了注释(表S7)。根据注释的分类信息,我们基于MetaTrass组装的高质量基因组构建了分类树,并在左侧直方图中附上每个基因组相应的N50值(图5)。图5中间的热图上则标注了通过传统策略获得的高质量基因组。MetaTrass组装结果对应的分类树的拓扑结构有助于人们全面了解微样本中生物的组成结构。从图5和图S9-S11来看,针对样本H_Gut_Meta01、H_Gut_Meta02、H_Gut_Meta03和P_Gut_Meta01,MetaTrass组装的高质量基因组所属目等级的数目分别为9、11、7和7。值得注意的是,一些目等级包含5个以上的高质量基因组,这将有助于人们研究同一样本中近缘微生物在全基因组尺度的遗传规律。对于样本H_Gut_Meta01(图5),有27个高质量的基因组被归入Lachnospirales目和14个被归入Oscillospirales目。根据分类分箱的丰度分布,这两个目正是该样本中的主要菌群。另外两个健康样本也得到了类似的结果(图S9和S10),说明测序覆盖率较高的微生物更有可能被MetaTrass组装出来。相比之下,对于P_Gut_Meta01来说,具有5个以上高质量基因组的目是肠杆菌目和放线菌目(图S11)。以上不同样本之间的差异与分类分箱结果中观察到的微生物组成差异一致。在测试中,MetaTrass成功组装了所有传统策略中的大部分高质量基因组。所有传统策略总共组装了137个不同的高质量基因组,其中只有25个没有被MetaTrass组装出来(图5)。热图结果显示,大多数传统策略的组装结果对应的目层级数与MetaTrass相似,但是每个目层级下的基因组数相对较少。比如, Supernova和MetaBAT2组合最多只在Lachnospirales目下组装了6个高质量基因组。此外,在MetaTrass组装的179个高质量基因组中,有146个基因组的N50大于100kb,这表明它提升组装连续性的性能也非常优异。
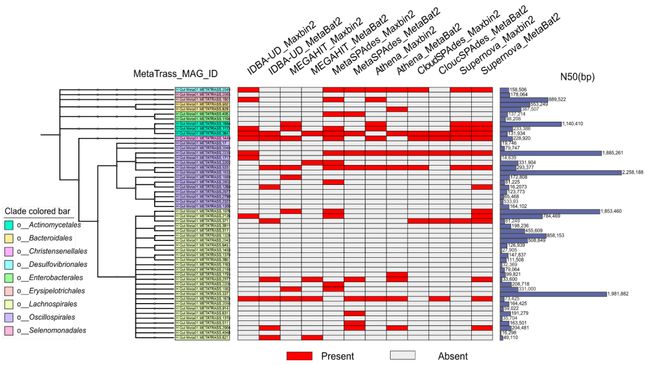
图5. H_Gut_Meta01样本中MetaTrass组装的高质量基因组构成的分类树
左图为分类树。中间的热图显示其它方法组装的高质量基因组。右侧的直方图是MetaTrass组装的高质量基因组的N50。
不同样本中的遗传差异分析
肠道微生物的不同类型基因变异与宿主的健康密切相关,研究人员通过研究不同微生物之间的遗传多样性来揭示不同地域或健康状况的人之间表型差异与微生物基因组的关联。在此,我们通过将基因组草图与参考基因组进行比对全面识别了不同样本中高质量基因组的菌种变异,包括单核苷酸变异(SNV)以及小的和大的插入缺失变异。对于不同类型的变异,四个样本的SNVs数量明显大于插入缺失变异的数量(图S12)。同时,三个健康样本中不同类型变异数的中位值相似,但其明显大于病人样本的中位值。根据QUAST的评估结果,我们发现病人样本中较小的变异数是由于其包含的基因组与参考基因组的差异远大于健康样本中的组装结果。然而,通过计算SNV密度来消除总比对长度的影响时,患者样本显示出比健康人样本更高的SNV密度(图S12)。病人样本的SNV密度中位值约为21,但健康人样本的中位值约为9。这种差异可能与个人的生理状态有关,而生理状态又与疾病、地域和种族相关。
根据高质量基因组的注释信息,我们发现有15个菌种同时出现在3个样本中,其中14个菌种出现在三个健康样本中,一个菌种出现在病人和两个健康样本中。通过分析三个健康样本中每个菌种的SNV密度和不同样本间的变异交集,我们进一步考察了不同样本中菌种间的遗传多样性。在同一个样本中,不同菌种的SNV密度也是不同的,但在不同的样本中同一菌种的SNV密度是相似的(图6A)。从图6B到6D,不同类型的样本独享和共享变异的数量对于不同的菌种有明显的波动,但它们在样本之间的差异表现出很大的一致性。H_Gut_Meta01和H_Gut_Meta02共享变异数是所有样本对中最多的。此外,三个健康样本共有的大尺度插入缺失变异的比例远远小于SNVs和小插入缺失变异。这些结果表明大变异比小变异更具有样本特异性,这与研究人员观察到的宿主健康与人类肠道微生物的大尺度结构变异更相关是一致的。
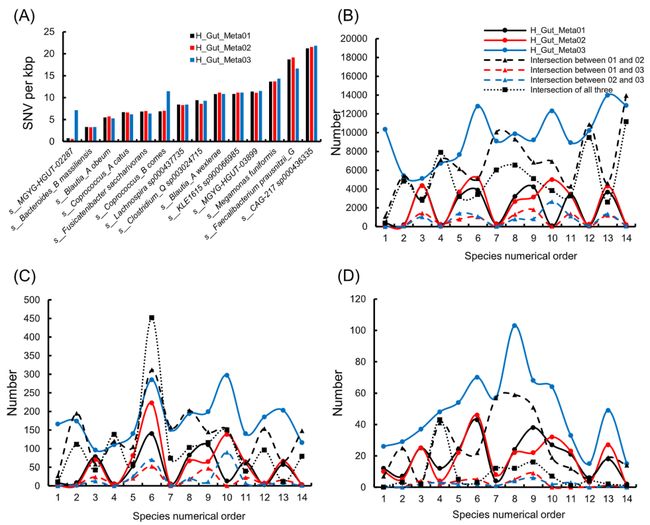
图6.单核苷酸变异(SNV)密度、独享变异及三个健康人样本共享变异的数量
(A) 图为SNV密度。(B)、(C)和(D)分别为SNVs、小和大的独享和共享变异的数量。子图(B)、(C)和(D)中的菌种数字序号对应于子图(A)中菌种从左到右的顺序
计算性能
我们记录了所有人类肠道数据集的运行时间和每个组装工具所使用的线程数(表1)。由于Athena和Supernova对内存的需求较大,除了它们是在高性能计算集群上进行测试之外,其它组装工具都在一台由24个英特尔(R)至强(R)银4116 CPU @ 2.10GHz的小型服务器上进行的测试。对于不同的样本,每个组装工具都设置了相同的线程数。表中的时间并没有包括从stLFR读长到10X链接读长的格式转换所消耗的时间。对于一个50Gb的数据集,该格式转换在单线程运行的情况下大约需要500分钟。我们的结果表明MetaTrass比Athena耗时少,但比其它组装工具耗时多。这可能是因为MetaTrass和Athena都包含许多子组装过程,实际上子组装是MetaTrass所有子过程中耗时最多的(表S8)。由于子组装过程是独立的,我们可以通过增加并行数来进一步提高MetaTrass的运行速度。在四个人类肠道微生物样本的测试中,默认子组装并行数为8,MetaTrass使用的内存峰值大于50Gb但小于100Gb。
表1 不同组装工具在四个人类肠道微生物样本数据集上的运行时间和所用线程数
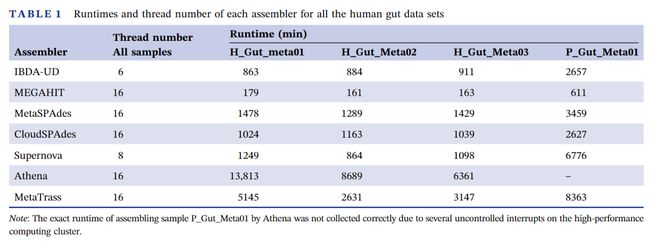
注:由于在高性能计算集群上组装样本P_Gut_Meta01数据集时,Athena出现了几个不受控的中断,其确切运行时间没有收集
讨 论
为了研究与人类肠道微生物群落有关的疾病的遗传根源,研究人员需要在菌种水平上获取大量的高质量菌种级基因组。然而,由于宏基因组组装存在着种间重复和丰度不均等高复杂度问题,如何在单个样本中获得足够数量的高质量基因组仍然是一个巨大的挑战。在该工作中,我们通过综合共标签信息和公共的微生物参考基因组开发了组装工具MetaTrass,它能获得具有精细分类分辨率的高质量基因组。与传统策略相比,MetaTrass在人类肠道微生物样本数据集上组装出了大量的高质量基因组。同时,MetaTrass也得到了大量高连续性基因组草图,其NG50值大于1 Mb。其中一些甚至比参考基因组的连续性还要好。对于所有四个人类肠道微生物样本,MetaTrass共组装了178个具有高完整度和低污染度的基因组草图,但其相对于参考基因组的覆盖度较低。MetaTrass组装的样本基因组与参考基因组之间的差异表明共标签信息可以减少读长分类分箱中的假阴性比率。通过共标签信息提供的关联,从种间重复和样本基因组特异性区域精炼得到的读长可以显著提高组装的质量。对于患者样本来说,MetaTrass组装的具有长连续性的高质量基因组的数量明显多于未执行共标签读长精炼所组装的基因组数目(图S13)。
MetaTrass最初是针对stLFR测序数据设计的,但也适用于其它类型的共标签测序数据。我们测试并分析了一个包含20个菌种的10X Genomics链接读长数据集ATCC Mock-20。该数据集有两个属其对应的菌种数大于一。我们的结果展示MetaTrass组装出了所有这些菌种的高质量基因组,而传统策略只获得了几个具有不同缺失度、异质性高和污染度高的基因组(图S14)。该结果表明MetaTrass能够在菌种或菌株等分辨率上组装宏基因组的潜力。需要注意的是,在该数据的共标签组装过程中,我们改用了CloudSPAdes,而非Supernova,因为部分菌种无法被Supernova成功组装。这可能由于链接读数中具有相同标签的长片段的数量大于stLFR数据,共标签读长精炼后的的数据集将包含更多的假阳性读长。
共标签信息的质量和共标签组装算法决定了MetaTrass的效率。其中共标签信息质量对共标签读长精炼的精度和灵敏度有很大影响。通过比较P_Gut_Meta01样本中具有中等丰度的菌种的不同读长集的基因组覆盖情况,我们评估了共标签读长精炼的敏感性。比较的数据集包括方法部分中定义的TR(分类分箱读长集)、共标签精炼读长集和所有读长集(表S10)。我们的结果显示精炼读长集中具有高深度覆盖区域的占比高于分类分箱读长集,但仍低于所有读长集。该结果表明共标签精炼后仍存在部分假阴性读长,它们可能由长片段的低覆盖率或不够长所导致的。在四个人类肠道微生物样本数据集的测试中,共标签组装工具Supernova消耗了绝大部分的计算时间;而且在链接读长数据集的测试中,Supernova也不能有效地组装具有高假阳性率的读长数据集。因此,对共标签文库技术和共标签读长精炼和组装算法的改进将有效地提升MetaTrass的性能。
结 论
总体而言,MetaTrass在人类肠道微生物样本中的应用显示了其在复杂微生物群落中组装出高质量且具有高分辨率的微生物基因组的巨大潜力。随着不同微生物群落参考基因组数量的增加和共标签测序技术的发展,MetaTrass所采用的先读长分类分箱后基因组组装的策略将得到进一步加强,同时将推动不同微生物群落中宿主表型与微生物基因型关联的研究
引文格式:
Yanwei Qi, Shengqiang Gu, Yue Zhang, Lidong Guo, Mengyang Xu, Xiaofang Cheng, Ou Wang, Ying Sun, Jianwei Chen, Xiaodong Fang, Xin Liu, Li Deng, and Guangyi Fan. 2022. MetaTrass: a high-quality metagenome assembler of the human gut microbiome by cobarcoding sequencing reads. iMeta e46.
https://doi.org/ 10.1002/imt2.46
代码和数据可用性
MetaTrass是开源的命令行应用程序,其在GitHub上的开源地址是https://github.com/BGI-Qingdao/MetaTrass,其满足开源协议General Public License v3.0。人工模拟样本和四个人类肠道微生物样本的组装结果和清洗后的stLFR数据集已被提交到公开数据库CNGBdb的CNSA(https://db.cngb.org/cnsa/)。该数据的登记号为CNP0002163。该数据属于受控数据类型,研究者可向CNGBdb的提出合理的申请而获得。Mock-20的10X Genomics链接读长数据集下载自网站https://trace.ncbi.nlm.nih.gov/Traces/index.html?run=SRR6760785。所有的补充材料(文本、图、表、中文翻译版本或视频)也可从相应的DOI或iMeta科学http://www.imeta.science/网站上获取
作者简介

齐彦伟(第一作者)
● 青岛华大基因研究院生物信息副研究员
● 本科毕业于安徽医科大学生物医学工程专业。主要研究方向为多组学应用研究和生物信息工具算法开发。在iMeta, Journal of Cancer,Current Genomics,Frontiers in Physiology等期刊发表SCI论文10余篇,以第一或共一作论文发表4篇,已申请专利5项,软件著作权7项

古盛强(第一作者)
● 中国科学院生命科学学院硕士。目前研究方向为微生物检测,精准医疗,大健康产业方向

邓礼(通讯作者)
● 青岛华大基因研究院生物信息副研究员
● 目前关注于新测序技术在基因组,宏基因组,单倍型分型等方向的算法研发。近五年以来,主持并结题国家自然科学基金1项,负责青岛华大基因研究院内部战略项目1项;在iMeta, GigaScience, Bioinformatics等期刊发表SCI论文6篇,其中第一或通讯作者论文4篇;申请专利4项,获批1项;申请并获批软件著作权4项

范广益(通讯作者)
● 青岛华大基因研究院主任科学家,研究员,中国科学院大学研究生导师
● 一直从事基因组学方面的工作,集中方向在动植物基因组的生物信息学工具开发和大数据挖掘,包括基因组De novo组装,群体遗传学,转录组及细胞组学等方面。通过综合运用实验、生物、数学及计算机知识,来挖掘动植物基因组有重要科学研究价值和应用的信息。主导和设计万种鱼类基因组、万种软体动物基因组和海洋哺乳类基因组学等多个重大项目,在《自然》、《科学》、《细胞》等国际知名学术刊物上发表科学论文100余篇,其中以第一作者或通讯作者(含共同)发表的论文30余篇,累积引用次数8000余次。主持和参与国家863计划、国家重点研发计划等国家及省部级项目9项,主持国家自然科学基金面上项目1项。担任Molecular Biology and Evolution等国际知名学术杂志审稿人
更多推荐
(▼ 点击跳转)
iMeta文章中文翻译+视频解读
iMeta封面 | 宏蛋白质组学分析一站式工具集iMetaLab Suite(加拿大渥太华大学Figeys组)

▸▸▸▸
iMeta | 成都中医药大学张杨组开发抗新冠中医药及其机制与疗效数据库
▸▸▸▸
iMeta | 深圳先进院戴磊组开发可同时提取共存菌株的组成和基因成分谱的菌株分析工具

▸▸▸▸
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法
▸▸▸▸
iMeta | 林雁冰/James M. Tiedje/谷洁等揭示菌群对寄生植物列当的调控作用

▸▸▸▸
iMeta | 华南农大陈程杰/夏瑞等发布TBtools构造Circos图的简单方法

▸▸▸▸
iMeta | 东农吴凤芝/南农韦中等揭示生物炭抑制作物土传病害机理
▸▸▸▸
iMeta | 叶茂/时玉等综述环境微生物组中胞内与胞外基因的动态穿梭与生态功能
▸▸▸▸
iMeta | 南农沈其荣团队发布微生物网络分析和可视化R包ggClusterNet

▸▸▸▸
iMeta | 华南师大王璋组综述人体肺部微生物组与人类健康和疾病之间的隐秘关联

▸▸▸▸
iMeta | 南科大夏雨组纳米孔测序揭示微生物可减轻高海拔冻土温室气体排放
期刊简介

“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:[email protected]
微信公众号
iMeta
责任编辑
微微