我们已经下载到了数据,在rawdata文件夹,总共1207个
1.文件合并
每个文件夹里面都有一个文本文件,现在需要把他们批量读入R语言,先分解一下,新建文件夹data_in_one,先把所有文本放在一个文件夹里面。
如果是用的命令行,就一行
cp rawdata/*/*.txt data_in_one
如果用R语言,这个for循环可以搞定。
每一个R语言初学者都应该掌握for循环!
### 使用for循环来批量做,回顾for循环的要点
for (dirname in dir("rawdata/")){
## 要查看的单个文件夹的绝对路径
mydir <- paste0(getwd(),"/rawdata/",dirname)
##找到对应文件夹中的文件并提取名称,pattern表示模式,可以是正则表达式
file <- list.files(mydir,pattern = "*.isoforms.*")
## 当前文件的绝对路径是
myfile <- paste0(getwd(),"/rawdata/",dirname,"/",file)
#复制这个文件到目的文件夹
file.copy(myfile,"data_in_one")
}
现在看看data_in_one里面是这样的。
2.构建读取函数
打开其中一个,检查并分析一下:

这个数据,我们只需要两列,一列是counts数,一列是有
MIMAT标志的那一列。用R语言读入是没有问题的,但是我们看到有重复行,这里我选择相同ID下的最大值来做下游分析。这时候,我可以写一个函数来做着一系列事情。
### 1.存储文件读入的顺序
files <- dir("data_in_one")
## 构建函数,读入数据,按照miRNA合并,选取最大counts
library(dplyr)
library(tidyr)
myfread <- function(files){
data.table::fread(paste0("data_in_one/",files)) %>%
separate(miRNA_region,into = c("stage","ID"),sep = ",") %>%
filter(stage == "mature") %>%
group_by(ID) %>%
summarise_at("read_count",max)
}
测试一下速度和效果
system.time(test1 <- myfread(files[1]))
system.time(test2 <- myfread(files[2]))
system.time(test3 <- myfread(files[3]))
读入的数据就两列,没问题:
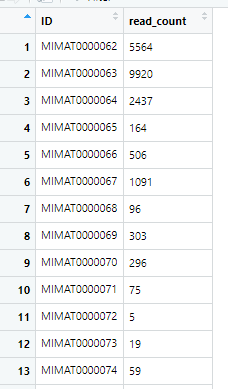
但是每次读入的行数不一样

这就是这里的细节,每个文件含有的有效miRNA不一样,所以最终合并的时候,不能使用cbind或者merge,只能兼容合并,就是full_join

3.找出样本名称注释文件
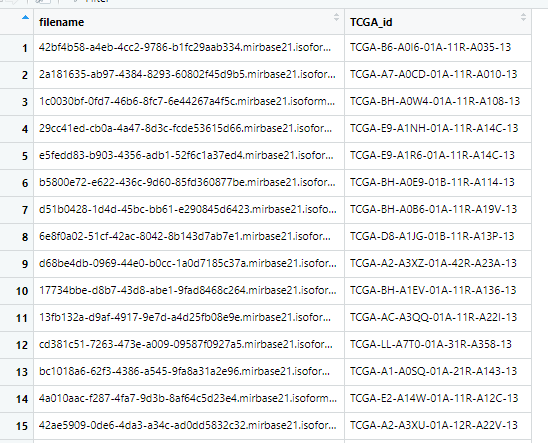
还有一个比较困难的问题在于,这些文本的名称很复杂,不是我们熟知的TCGA_id, 所以还需要找到一个文件名称和TCGA_id对应的文件,要不然读入R语言后,分不清样本信息了,这个文件可以从下载的metadata文件中获取。
metadata <- jsonlite::fromJSON("metadata.cart.2019-08-14.json")
library(dplyr)
metadata_id <- metadata %>%
dplyr::select(c(file_name,associated_entities))
## 提取对应的文件
naid_df <- data.frame()
for (i in 1:nrow(metadata)){
naid_df[i,1] <- metadata_id$file_name[i]
naid_df[i,2] <- metadata_id$associated_entities[i][[1]]$entity_submitter_id
}
colnames(naid_df) <- c("filename","TCGA_id")
这样,对应关系就出来了。
4.批量读入数据
lapply配合自定义函数,我们可以无所不能
f <- lapply(files,myfread)
下面这一步,用Reduce函数实现多参数批量操作,就是把每一个样本数据都兼容合并
Reduce函数实现多个数据框批量合并
f <- Reduce(function(x,y) dplyr::full_join(x = x, y = y, by = "ID"),f)
f <- as.data.frame(f)

现在数据是2236行。马上开始样本名称转换。
5.样本名称转换
先把第一列变成行名,再把第一列删掉
rownames(f) <- f[,1]
f <- f[,-1]
因为机器看到的第一个样本跟我们看到的第一个样本不一样,所以我们要用机器读入的样本顺序(存储在了files中)去调整对应关系中的TCGA_id
rownames(naid_df) <- naid_df[,1]
naid_df <- naid_df[files,]
现在可以命名了
colnames(f) <- naid_df$TCGA_id
6.数据预处理
读入的数据,因为是兼容合并的,所以空缺都用NA来表示,现在我们要把他们转换为0
f[is.na(f)] <- 0
然后,我要把行名变成第一列,这里我特别喜欢使用的是cbind
expr_df <- cbind(ID=rownames(f),f)

最后要保存一下数据为Rdata格式
save(expr_df,file = "BRCA_miRNA_seq.Rdata")
今天的内容到这里,明天会将下游分析啊。
在这个系列结束后,我还会录制视频帮助理解,会发布在果子学生信公众号,到时候通知大家。