转录组学习一(软件安装)
转录组学习二(数据下载)
转录组学习三(数据质控)
转录组学习四(参考基因组及gtf注释探究)
转录组学习五(reads的比对与samtools排序)
转录组学习六(reads计数与标准化)
转录组学习七(差异基因分析)
转录组学习八(功能富集分析)
任务
- 载入表达矩阵,然后设置好分组信息
- 用DEseq2进行差异分析,也可以走走edgeR或者limma的voom流程
- 基本任务是得到差异分析结果,进阶任务是比较多个差异分析结果的异同点。
- 了解差异基因分析背后的统计学基础(数据标准化,假设检验)
差异基因分析背后的统计学知识
简单回顾之前genek-转录组原理篇的知识点,参考学习徐洲更的转录组分析流程七(差异基因分析),及相关统计学书本知识。
一:样品间(组间的数据)的表达标准化
- (处理样品中,有一个基因的表达量异常的高,大量的reads被占,导致其他gene的相对表达量下降。)TPM的标准化是在 各自的==样品内部==的 差异基因==相对表达量==对于组间来说,也就是处理组和对照组来说就需要进一步的标准化。目的就是能够得到==样品间的绝对表达量==是否有差异。
- 标准化方法:
- 内参基因,看家基因。某一基因在样本中的表达肯定是一样的。(有限制,依赖基因的功能注释,数目少准确不高。)
- 通用方法:假设大多数基因是没有差异表达的。统计学方法找到标准化的因子。
- DESeq2(estimateSizeFactors/sizeFactors)、edgeR(TMM,calcNormFactors)。差异表达分析鉴定软件。
- 输入文件是reads count矩阵。
- 可用Trinity软件包里的run_DE_analysis.pl
- FPKM/TPM/TMM的选择
- 组内比较 TPM(不同基因在同一样品中的表达差异)
- 组间比较 TMM(同一基因在不同样品间的表达差异)
- 低表达基因的过滤TMM-normalized FPKM(用于数据过滤、heatmap、共表达等)
- TMM-TPM还未结合起来(可能TMM矫正TPM后意义就变了?或者TMM还未流行起来)
二:假设检验进行差异表达基因的鉴定
- 建库测序是一个随机抽样的过程。纵使是两次测序,即抽样,同一基因的reads_count也会有差异。
- 当观察到的基因在两组reads_count 中不一样的时候,可能有两种原因:可能是==表达丰度上的差异,也可能是随机抽样过程中的波动==。假设是随机波动导致的,然后计算在随机波动的前提下,出现当前事件的概率,如果概率低。 反证法。
- 假设检验:需要根据实际情况(总体均值、方差是否已知,单样本还是多样本,重复个数的多少/大样本量还是小样本量)
 image
image - 差异表达基因通常用t检验(组间差异除以/组内差异,组间差异更大,而组内差异小则更可能有差异【此即是需要测更多生物学重复,能更准确的估计组内差异】)
- t值的双侧检验和单侧检验。
- 单侧检验:G1在处理组中表达 大于 对照组。右侧面积->则单侧检验,即upregulate。
- 双侧检验:G1处理组与对照组有明显差异。则双侧检验
- 一类错误(即为假阳性,与P-value对应,0.05 or 0.01),二类错误(指漏诊率)
 image
image- 降低一类错误:对于一条gene可能有5%可能误诊,而在差异基因鉴定时都是大规模的,如果一万条,50条则太高了。p-value 进一步的矫正 FDR方法,q-value为升级版。
- 降低二类错误:G3增加生物学重复,G2增加测序量来提高抽样次数。
 image
image
用DESeq2进行差异表达分析
一:DESeq2的安装
source("https://bioconductor.org/biocLite.R") 载入安装工具bioconductor
biocLite("DESeq2") bioconductor安装DESEQ2
library(DESeq2)
二:DESeq2差异分析基本流程
- 对于DESeq2需要输入的三个数据:表达矩阵、样品信息矩阵、差异比较矩阵
- 而对于DESeq2的差异分析步骤,总结起来就是三步:
- 构建一个dds(DESeqDataSet)的对象;
- 利用DESeq函数进行标准化处理;
- 用result函数来提取差异比较的结果。
三:构建dds矩阵
- 构建dds矩阵的基本代码
dds <- DESeqDataSetFromMatrix(countData = cts, colData = coldata, design= ~ batch + condition)
-
其输入的三个文件:
- 表达矩阵:即代码中的countData,就是通过之前的HTSeq-count生成的reads-count计算融合的矩阵。行为基因名,列为样品名,值为reads或fragment的整数。
- 样品信息矩阵:即上述代码中的colData,它的类型是一个dataframe(数据框),第一列是样品名称,第二列是样品的处理情况(对照还是处理等)。可以从表达矩阵中导出或是自己单独建立。
- 差异比较矩阵:即代码中的design,差异比较矩阵就是告诉差异分析函数哪些是对照,哪些是处理。
正式构建dds的矩阵:
输入HTSeq-count的各个结果文件
KD1 <- read.table("mus_E14_cell_Akap95_shRNA_rep1_SRR3589959.count",sep = "\t",col.names = c("gene_id","rep1"))
KD2 <- read.table("mus_E14_cell_Akap95_shRNA_rep2_SRR3589960_matrix.count",sep = "\t",col.names = c("gene_id","rep2"))
control1 <- read.table("mus_E14_cell_control_shRNA_rep1_SRR3589961_matrix.count",sep = "\t",col.names = c("gene_id","control1"))
control2 <- read.table("mus_E14_cell_control_shRNA_rep2_SRR3589962_matrix.count",sep = "\t",col.names = c("gene_id","control2"))
- merge_file
raw_count <- merge(merge(merge(KD1,KD2,by="gene_id"),control1,by="gene_id"),control2,by="gene_id")
- 构建DESeq_data_set基本信息
count_data <- raw_count[,2:5]
row.names(count_data) <- raw_count[,1]
condition <- factor(c("rep","rep","condition","condition"))
col_data <- data.frame(row.names = colnames(count_data), condition)
dds <- DESeqDataSetFromMatrix(countData = count_data,colData = col_data,design = ~ condition)
- 过滤低质量的低count数据:
nrow(dds) ## 显示52636行
dds_filter <- dds[ rowSums(counts(dds))>1, ] ##将所有样本基因表达量之和小于1的基因过滤掉
nrow(dds_filter) ##显示27101行,过滤掉了近50%
四:对dds矩阵进行DESeq分析:
- 使用DESeq进行差异表达分析:(一堆统计统计学知识的坑待填啊)
- estimation of size factors(estimateSizeFactors),
- estimation of dispersion(estimateDispersons),
- Negative Binomial GLM fitting and Wald statistics(nbinomWaldTest)
-
DESeq自动化的基本过程如图:
 image
image
dds_out <- DESeq(dds_filter)
res <- results(dds_out)
summary(res)
- 结果显示: 2.6%的基因:717个上调,2.1%的基因579下调,另外还有41%的基因表达量是不高的,属于low count。
 image
image
五:提取差异基因分析的结果
- 在利用数据比较分析两个样品中同一个基因是否存在差异表达的时候,一般选取Foldchange值和经过FDR矫正过后的p值,取padj值(p值经过多重校验校正后的值)小于0.05,log2FoldChange大于1的基因作为差异基因集
table(res$padj<0.05)
res_deseq <- res[order(res$padj),]
diff_gene_deseq2 <- subset(res_deseq, padj<0.05 & (log2FoldChange > 1 | log2FoldChange < -1))
## or diff_gene_deseq2 <- subset(res_desq, padj<0.05 & abs(log2FoldChange) >=1)
diff_gene_deseq2 <- row.names(diff_gene_deseq2)
res_diff_data <- merge(as.data.frame(res),as.data.frame(counts(dds_out,normalize=TRUE)),by="row.names",sort=FALSE)
write.csv(res_diff_data,file = "11_30_mouse_data.csv",row.names = F)
-
提取结果:
 image
image
六 MA图
MA-plot (R. Dudoit et al. 2002) ,也叫 mean-difference plot或者Bland-Altman plot,用来估计模型中系数的分布。 X轴, the “A” ( “average”);Y轴,the “M” (“minus”) – subtraction of log values is equivalent to the log of the ratio。
M表示log fold change,衡量基因表达量变化,上调还是下调。A表示每个基因的count的均值。根据summary可知,low count的比率很高,所以大部分基因表达量不高,也就是集中在0的附近(log2(1)=0,也就是变化1倍).提供了模型预测系数的分布总览。
library(ggplot2)
plotMA(res,ylim=c(-5,5))
topGene <- rownames(res)[which.min(res$padj)]
with(res[topGene, ], {
points(baseMean, log2FoldChange, col="dodgerblue", cex=2, lwd=2)
text(baseMean, log2FoldChange, topGene, pos=2, col="dodgerblue")
})

- lfcShrink 收缩log2 fold change
resLFC <- lfcShrink(dds_out,coef = 2,res=res)
plotMA(resLFC, ylim=c(-5,5))
topGene <- rownames(resLFC)[which.min(res$padj)]
with(resLFC[topGene, ], {
points(baseMean, log2FoldChange, col="dodgerblue", cex=2, lwd=2)
text(baseMean, log2FoldChange, topGene, pos=2, col="dodgerblue")
})
idx <- identify(res$baseMean, res$log2FoldChange)

七:gene 聚类热图
基本上是按照PANDA姐的分析的代码走一遍,很多参数的含义还不是很清楚,等接下来的时间再学习作图相关。
library(genefilter)
library(pheatmap)
rld <- rlogTransformation(dds_out,blind = F)
write.csv(assay(rld),file="mm.DESeq2.pseudo.counts.csv")
topVarGene <- head(order(rowVars(assay(rld)),decreasing = TRUE),20)
mat <- assay(rld)[ topVarGene, ]
### mat <- mat - rowMeans(mat) 减去一个平均值,让数值更加集中。第二个图
anno <- as.data.frame(colData(rld)[,c("condition","sizeFactor")])
pheatmap(mat, annotation_col = anno)
结果两个热图:
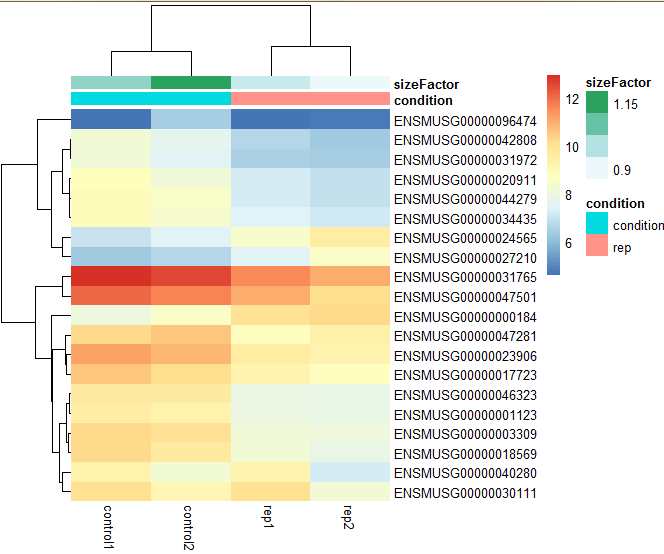

八:火山图
#### perl 单行:第三列log2foldchange大于1为上调,小于-1下调,其他的不显著。
perl -F'\t' -alne '$F[5]=~s/\r//;if(/baseMean/){print "gene\tlog2FoldChange\tpadj\tsignificant"} else{unless(/NA/){if($F[2] >=1 && $F[5]<0.05){print "$F[0]\t$F[2]\t$F[5]\tup"} elsif($F[2]<=-1 && $F[5]<0.05){print "$F[0]\t$F[2]\t$F[5]\tdown"} else{print "$F[0]\t$F[2]\t$F[5]\tno"}}}' mm_deseq2_resLFS.csv >volcano.txt
### 火山图
data <-read.table(file="volcano.txt",header = TRUE, row.names =1,sep = "\t")
volcano <-ggplot(data = volcano_data,aes(x=log2FoldChange,y= -1*log10(padj)))+geom_point(aes(color=significant))+scale_color_manual(values = c("red","grey","blue")) + labs(title="Volcano_Plot",x=expression((log[2](FC)), y=expression(-log[10](padj)) ))+geom_hline(yintercept=1.3,linetype=4)+geom_vline(xintercept=c(-1,1),linetype=4)
volcano

11/30下午小结:本次差异基因分析的学习,代码部分基本上是参考学习着各个大神们的笔记一步步操作来的。好的方面是总算对差异基因分析有了一定系统化的了解,对DESeq2差异分析软件了解了基本使用方法,对一些简单可视化图形的展示与解读有了一定的认识。但仍然有许多不足,许多坑待填比如:R语言,ggplot2可视化方面,还有最大的坑:统计学相关知识。这些将会是转录组学习完之后新的学习方向。
参考文章
- 【PANDA姐的转录组入门-差异基因分析-2】 https://mp.weixin.qq.com/s/05Gv1pnL0GWZwjz893xcOw
- 【用DESeq2包来对RNA-seq数据进行差异分析】http://www.bio-info-trainee.com/bioconductor_China/software/DESeq2.html
- 【(伪)从零开始学转录组(7):差异基因表达分析】https://mp.weixin.qq.com/s?__biz=MzI1MjU5MjMzNA==&mid=2247484514&idx=1&sn=1c6d227c6d7ac432d8baffb93b0b9072&chksm=e9e02dc3de97a4d59d918ee37655153fa4ccc8122e3259c0201ff441c922548f22f84311ad91&scene=21#wechat_redirect
- 【鉴定差异基因,哪家强?】https://mp.weixin.qq.com/s?__biz=MzI4NjMxOTA3OA==&mid=2247484152&idx=1&sn=b27809490589b6082ce1ea0cc3be878f&chksm=ebdf8a71dca803674a7865b41e63b3900d3bd5b257b46a1a89ddad1326fe784a19b3a732dfd0&scene=21#wechat_redirect
- 【青山屋主-转录组入门】https://zhuanlan.zhihu.com/p/30350531?group_id=905527998389362688
- 【Analyzing RNA-seq data with DESeq2
】http://www.bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html - 【转录组差异表达筛选的真相
】https://mp.weixin.qq.com/s?__biz=MzI4NjMxOTA3OA==&mid=2247484080&idx=1&sn=7de397add158271aa51763d0d3598deb&chksm=ebdf8a39dca8032f1cb8b8154a024ee7f564c2e33b2f824efce7e34a016ef7baf92d52d64c79&scene=21#wechat_redirect - 【FPKM / RPKM与TPM哪个用来筛选差异表达基因更准确?你可别逗了!】https://mp.weixin.qq.com/s?__biz=MzI4NjMxOTA3OA==&mid=2247483987&idx=1&sn=aa2ca81e7fe128edaaedc47479c517c9&chksm=ebdf8adadca803cc31261a1ccabf8a6bdb835ce12b670cc01969fcfde51cfdb991d0619e7695&scene=21#wechat_redirect