RHive基础函数
加载RHive
library(RHive)
在加载之前,首先要配置HADOOP_HOME和HIVE_HOME环境变量。
也可临时设置环境变量:
Sys.setenv(HIVE_HOME=”/service/hive-0.7.1”)
Sys.setenv(HADOOP_HOME=”/service/hadoop-0.20.203.0”)
library(RHive)
rhive.init环境变量配置后自动启动,或
rhive.init(hive='/path/to/hive',hadoop_home='/path/to/hadoop'[,hadoop_conf='/path/to/hadoop_conf'])
rhive.connect
rhiveConnection <- rhive.connect(“10.1.1.1”)rhive.query
hive支持SQL语句,特别和MySQL很像。rhive.query发送SQL语句到hive并从hive中接收结果。
例:resultDF <- rhive.query(‘SELECT * FROM usarrests’)
resultDF是一个data frame。
一个注意的事情:如果 抽取的数据超出RHive服务器内存或电脑的内存,将会出现错误。但是可以先创建一个临时表,把结果放入进去:
rhive.query(“
CREATE TABLE new_usarrests(
rowname string,
murder double,
assault int,
urbanpop int,
rape double
)”)
rhive.query(“INSERT OVERWRITE TABLE new_usarrests SELECT * FROM usarrests”)rhive.close
结束使用Hive,,不再使用RHive其他函数,则可断开连接
conn <- rhive.connect()
rhive.close(conn)rhive.list.tables
返回Hive中所有的表,同rhive.query(“SHOW TABELS”)一样
rhive.desc.table
描述所选择的表
rhive.desc.table(“usarrests”), 和rhive.query(“DESC usarrests”)一样。
rhive.load.table
和R的data.frame对象一样
df <- rhive.load.table(“usarrests”)
和df <- rhive.query(“SELECT * FROM usarrests”)相同
rhive.write.table
如果你想在Hive加入一张表,你必须首先创建一张表,但使用rhive.write.table,就不需要创建一张表了,直接把R的数据框插入Hive中就可以了。
rhive.write.table(UScrime), UScrime是R的一个数据框
如果Hive中表已存在,则会出现错误
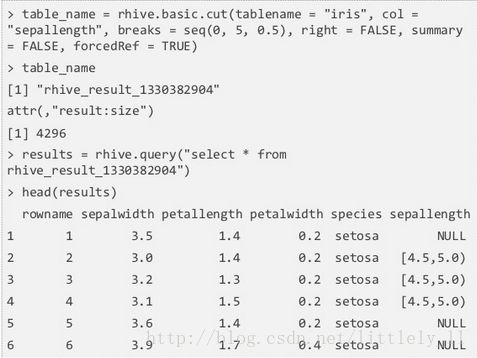
rhive.basic.cut
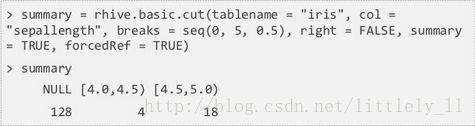
rhive.basic.cut把表中的数值列转化为因子列。首先将数值列的范围切分成区间,再把区间内的数值转化为因子。rhive.basic.cut接受6个参数,tablename,col,breaks,right,summary和forcedRef。breaks是数值列的切割点;right表示区间的开闭,TRUE为左开右闭,FALSE相反;summary=TRUE分割数值计数对应于各个区间,如果FALSE,返回一个由因子表更新的新表的表名;forcedRef=TRUE返回一个表名而不是数据框。
summary=FALSE:
summary=TRUE:
rhive.basic.xtabs
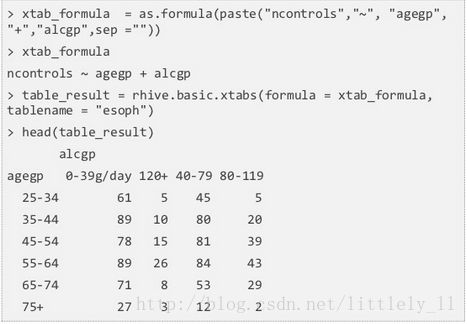
rhive.basic.xtabs从交叉分类因子中创建一个列联表,参数为一个formula对象和一个表名,返回一个矩阵形式的列联表。
esoph数据:
rhive.basic.t.test
rhive.basic.t.test应用Welch t检验,检验双样本均值差是否有差异,原假设为两样本均值差不等于0,双边检验。
下面检验了iris的sepal length和petal length的均值差,注意函数是怎样调用sepal length和petal length的。
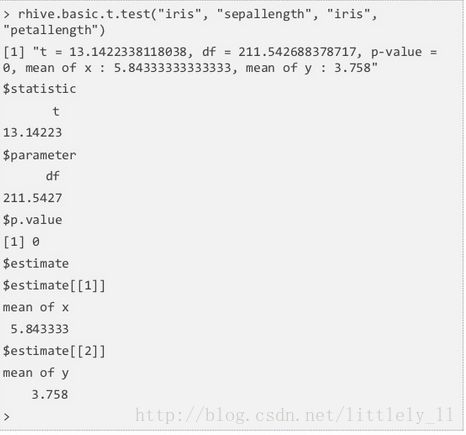
p值为0,接受备择假设,说明两者均值确有差异。
rhive.basic.scale
rhive.basic.scale把数值数据转化为0均值,标准差为1的数据。第一个参数为表名,第二个参数为输出的列名。
返回的列表中,增加了一个“scaled_column name”的列,并保存为字符串,它是可编辑的。

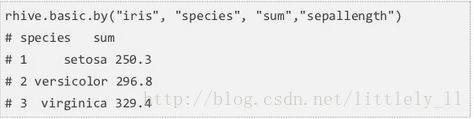
rhive.basic.by
rhive.basic.by把特定的列分组。
下面代码是对species分组(group by),然后对sepallength应用sum函数并返回结果。返回结果中会发现每一种类和sepallength的和。
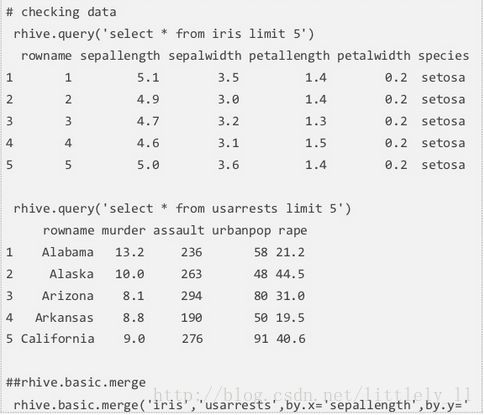
rhive.basic.merge
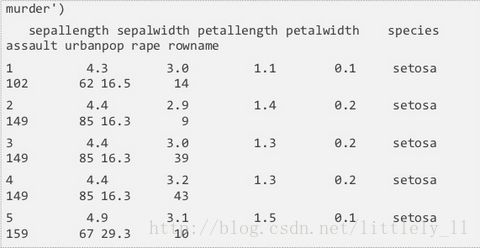
rhive.basic.merge依据相同的行融合两个表。类似SQL中的join。
rhive.basic.range
返回最大和最小值
![]()