环境准备
安装软件: 参考「基因组学」使用OrthoFinder进行直系同源基因分析 安装OrthoFinder,然后再安装CAFE
wget https://github.com/hahnlab/CAFE/releases/download/v4.2/CAFE-4.2.tar.gz
cd CAFE
./configure
make
make install
# 如果没有root权限
mkdir -p ~/opt/biosoft/CAFE-4.2/bin
make install prefix=~/opt/biosoft/CAFE-4.2
数据准备: 一共会分析mouse, rat, cow, horse, cat, marmoset,macaque, gibbon, baboon, orangutan, chimpanzee, 和 human 12个物种的蛋白数据。既可以在Ensemble上下载,也可以从 https://share.weiyun.com/5ZIjBg8 (密码:3jzdpm)下载。
解压缩
tar xf twelve_spp_proteins.tar.gz
for i in `ls -1 twelve_spp_proteins/*.tar.gz`; do tar xf $i -C twelve_spp_proteins; done
rm twelve_spp_proteins/*.tar.gz
识别基因家族
识别物种内和物种间的基因家族分为如下四步:
- 仅保留每个基因中有代表性的转录本,去除可变剪切和冗余基因
- 建立BLAST数据库,使用blastp进行 all-by-all 的比对
- 使用MCL基于blastp结果进行聚类,基因序列相似的通常是一个基因家族
- 解析MCL的输出结果,用作CAFE的输入
将所有最长的转录本合并成单个文件
提取每个基因中最长的转录本,然后合并成单个文件。
python python_scripts/cafetutorial_longest_iso.py -d twelve_spp_proteins/
cat twelve_spp_proteins/longest_*.fa | seqkit rmdup - > makeblastdb_input.fa
All-by-all BLAST
先用makeblastdb建立BLAST数据库
makeblastdb -in makeblastdb_input.fa -dbtype prot -out blastdb
之后用blastp进行序列搜索,得到每个序列的相似序列
blastp -num_threads 20 -db blastdb -query makeblastdb_input.fa -outfmt 7 -seg yes > blast_output.txt &
-seg参数过滤低复杂度的序列(即氨基酸编码为X), -num_threads线程数,此处设置为20.
这一步运行的时间和你服务器性能有关,你可以读完后续的内容,再继续分析。
使用MCL进行序列聚类
现在,我们需要根据BLAST输出中序列相似性信息寻找聚类。这些聚类将是后续用于CAFE分析的基因家族。聚类这一步将通过mcl处理。
使用shell命令将BLAST转成MCL能够识别的ABC格式
grep -v "#" blast_output.txt | cut -f 1,2,11 > blast_output.abc
下一步,创建网络文件(.mci)和字典文件(.tab),然后进行聚类
mcxload -abc blast_output.abc --stream-mirror --stream-neg-log10 -stream-tf 'ceil(200)' -o blast_output.mci --write-tab blast_output.tab &
# --stream-mirror: 为输入创建镜像,即每一个X-Y都有一个Y-X
# --stream-neg-log10: 对输入的数值做-log10 转换
# -stream-tf: 对输入的数值进行一元函数转换,这里用到的是ceil(200)
根据mci文件进行聚类, 其中主要调整的参数是-I, 它决定了聚类的粒度,值越小那么聚类密度越大,这个值没有想象中的那么至关重要。一般设置为3,你也可以尝试用其他值,然后比较结果。最终的目的是正确分析物种间的直系同源基因。
mcl blast_output.mci -I 3
mcxdump -icl out.blast_output.mci.I30 -tabr blast_output.tab -o dump.blast_output.mci.I30
整理MCL的输出结果
上一步MCL的输出还不能直接用于CAFE,还需要对其进行解析并过滤。
第一步是将原来的mcl格式转成CAFE能用的格式
python python_scripts/cafetutorial_mcl2rawcafe.py -i dump.blast_output.mci.I30 -o unfiltered_cafe_input.txt -sp "ENSG00 ENSPTR ENSPPY ENSPAN ENSNLE ENSMMU ENSCJA ENSRNO ENSMUS ENSFCA ENSECA ENSBTA"
这里的"ENSG00" 是ENSEMBL编号中物种的标识符。并且标识符之前只能有一个空格,否则输出结果就是下面这个情况
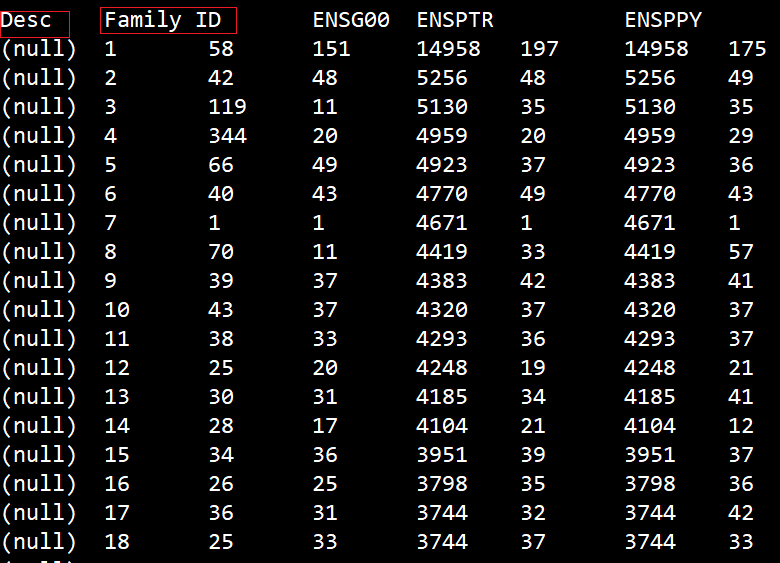
正确的输出结果应该是如下所示

第二步,将那些基因拷贝数变异特别大的基因家族剔除掉,因为它会造成参数预测出错。下面的脚本是过滤掉一个或多个物种有超过100个基因拷贝的基因家族,虽然不是特别的严格,但效果和根据拷贝数变异过滤类似
python python_scripts/cafetutorial_clade_and_size_filter.py -i unfiltered_cafe_input.txt -o filtered_cafe_input.txt -s
将原本的编号替换成有意义的物种名
sed -i -e 's/ENSPAN/baboon/' -e 's/ENSFCA/cat/' -e 's/ENSBTA/cow/' -e 's/ENSNLE/gibbon/' -e 's/ENSECA/horse/' -e 's/ENSG00/human/' -e 's/ENSMMU/macaque/' -e 's/ENSCJA/marmoset/' -e 's/ENSMUS/mouse/' -e 's/ENSPPY/orang/' -e 's/ENSRNO/rat/' -e 's/ENSPTR/chimp/' filtered_cafe_input.txt
sed -i -e 's/ENSPAN/baboon/' -e 's/ENSFCA/cat/' -e 's/ENSBTA/cow/' -e 's/ENSNLE/gibbon/' -e 's/ENSECA/horse/' -e 's/ENSG00/human/' -e 's/ENSMMU/macaque/' -e 's/ENSCJA/marmoset/' -e 's/ENSMUS/mouse/' -e 's/ENSPPY/orang/' -e 's/ENSRNO/rat/' -e 's/ENSPTR/chimp/' large_filtered_cafe_input.txt
物种树推断
构建物种树主要分为多序列联配和系统发育树推测两步, 之后在已有进化树的基础上构建超度量树用作CAFE输入。
多序列联配一般用的是单拷贝的直系同源基因,分别进行多序列联配之后然后合并成单个文件。接着用系统发育树推测软件进行建树,可选软件有
- 极大似然法: RAxML, PhyML, FastTree
- 贝叶斯法: MrBayes
这里不展示具体过程,直接用已有的极大似然树的结果(NEWICK格式),保存为twelve_spp_raxml_cat_tree_midpoint_rooted.txt
(((cow :0.09289 ,( cat :0.07151 , horse :0.05727) :0.00398) :0.02355 ,(((( orang:0.01034 ,( chimp :0.00440 , human :0.00396) :0.00587) :0.00184 , gibbon:0.01331) :0.00573 ,( macaque :0.00443 , baboon :0.00422) :0.01431):0.01097 , marmoset :0.03886) :0.04239) :0.03383 ,( rat :0.04110 , mouse:0.03854) :0.10918);

推断超度量树
超度量树(ultrametric tree)也叫时间树,就是将系统发育树的标度改成时间,从根到所有物种的距离都相同。构建方法有很多,比较常用的就是r8s.
这里用cafetutorial_prep_r8s.py构建r8s的批量运行脚本,然后提取超度量树
python python_scripts/cafetutorial_prep_r8s.py -i twelve_spp_raxml_cat_tree_midpoint_rooted.txt -o r8s_ctl_file.txt -s 35157236 -p 'human,cat' -c '94'
r8s -b -f r8s_ctl_file.txt > r8s_tmp.txt
tail -n 1 r8s_tmp.txt | cut -c 16- > twelve_spp_r8s_ultrametric.txt
运行CAFE
运行CAFE有两种模式,一种是CAFE的命令行模式,先执行cafe进行CAFE的shell, 然后在其中执行命令。另一种是脚本模式,也就是你先把命令编辑完成,然后用cafe script_to_run.sh运行。
估计出生-死亡参数
CAFE的主要功能就是根据给定的进化树和基因家族数估计一个或多个 birth-death()参数。 参数描述的是基因出现或者消失的概率。
为整个树估计单个
编辑cafetutorial_run1.sh。CAFE的命令不能有额外的空格出现在 tree后面的()中,以及lambda 的 -t 后的()中,否则运行时会无法正确解析文件导致报错。
#!cafe
load -i filtered_cafe_input.txt -t 4 -l reports/log_run1.txt
tree ((((cat:68.710507,horse:68.710507):4.566782,cow:73.277289):20.722711,(((((chimp:4.444172,human:4.444172):6.682678,orang:11.126850):2.285855,gibbon:13.412706):7.211527,(macaque:4.567240,baboon:4.567240):16.056992):16.060702,marmoset:36.684935):57.315065):38.738021,(rat:36.302445,mouse:36.302445):96.435575)
lambda -s -t ((((1,1)1,1)1,(((((1,1)1,1)1,1)1,(1,1)1)1,1)1)1,(1,1)1)
report reports/report_run1
然后运行如下命令
mkdir -p reports
cafe cafetutorial_run1.sh
结果统计
上一步运行结束后的报告文件在reports/reportrun1.cafe,可以用已有的脚本分析哪些基因家族发生了扩张或者搜索
python python_scripts/cafetutorial_report_analysis.py -i reports/report_run1.cafe -o reports/summary_run1
在reports文件夹下会出现如下文件
-
summary_run1_node.txt: 统计每个节点中扩张,收缩的基因家族数 -
summary_run1_fams.txt: 具体发生变化的基因家族
为高基因拷贝数的基因家族预测
之前过滤掉的高拷贝数变异的基因家族可以单独进行分析, 运行命令如下
#!cafe
load -i large_filtered_cafe_input.txt -t 4 -l reports/log_run2.txt
tree ((((cat:68.710507,horse:68.710507):4.566782,cow:73.277289):20.722711,(((((chimp:4.444172,human:4.444172):6.682678,orang:11.126850):2.285855,gibbon:13.412706):7.211527,(macaque:4.567240,baboon:4.567240):16.056992):16.060702,marmoset:36.684935):57.315065):38.738021,(rat:36.302445,mouse:36.302445):96.435575)
lambda -l 0.00265952 -t ((((1,1)1,1)1,(((((1,1)1,1)1,1)1,(1,1)1)1,1)1)1,(1,1)1)
report reports/report_run2
为树的不同部分预测多个
如果你认为不同物种或者不同分支的基因家族进化速率不同,那么可以让CAFE预测多个值. 对lambda部分进行调整, 相同数字表示相同,不同数字表示不同。
#!cafe
load -i filtered_cafe_input.txt -t 4 -l reports/log_run3.txt
tree ((((cat:68.710507,horse:68.710507):4.566782,cow:73.277289):20.722711,(((((chimp:4.444172,human:4.444172):6.682678,orang:11.126850):2.285855,gibbon:13.412706):7.211527,(macaque:4.567240,baboon:4.567240):16.056992):16.060702,marmoset:36.684935):57.315065):38.738021,(rat:36.302445,mouse:36.302445):96.435575)
lambda -s -t ((((3,3)3,3)3,(((((1,1)1,2)2,2)2,(2,2)2)2,3)3)3,(3,3)3)
report reports/report_run3
CAFE主要输出文件格式
CAFE主要输出内容如下,下游分析所需信息需要通过对其解析获得。

Lambda是整个进化树的预测值
# IDs of nodes表示不同节点的编号,这里cat为0,horse为2,cat和horse所在的节点是1.
最后是每个基因家族的结果。以最开始的表示行为例,第一列对应输入基因家族的编号;第二列是Newick的进化树,cat_59中的59表示该基因家族在cat里有59个基因;第三列是Family-wide P-value,用于表明该基因家族是否是显著性的扩张或是收缩,这里是0.438,说明变化不明显。在第三列的p值小于0.01时,第四列表明哪个分支的基因家族发生了变化,上图中只有ID 11的基因家族有变化, 但是0,1,2,4分支并没有变化。
参考资料
- https://iu.app.box.com/v/cafetutorial-pdf