Mechanisms of disease: The oxidative stress theory of diabetic neuropathy
疾病机制:糖尿病神经病变的氧化应激理论
Rev Endocr Metab Disord (2008) 9:301–314
文献DOI:10.1007/s11154-008-9104-2
文献PMID:18709457
文献原文链接:https://sci-hub.se/10.1007/s11154-008-9104-2

摘要:
糖尿病神经病变是糖尿病最常见的并发症,影响超过50%的糖尿病患者。目前,糖尿病神经病变的唯一治疗方法是血糖控制和细致的足部护理。在这篇综述中,我们讨论了过量葡萄糖超载电子传递链的观点,导致超氧化物的产生以及随后的线粒体和细胞溶质氧化应激。代谢和血管途径中的缺陷与氧化应激相交,以产生糖尿病性神经病中存在的神经损伤的发生和进展。这些途径包括晚期糖基化终产物的产生,山梨糖醇、己糖胺和蛋白激酶C途径的改变以及聚-ADP核糖聚合酶的活化。新的生物信息学方法可以增进当前的研究,并导致新的发现,以了解糖尿病神经病变的发病机制,并确定更有效的分子治疗目标。
关键词:糖尿病,神经病病变,氧化应激,生物信息学
1、简介
糖尿病性神经病变是糖尿病最常见的并发症。虽然根据诊断糖尿病神经病变的方法,估计值各不相同,但通常认为至少50%的糖尿病患者在其一生中会发生神经病变[1-7]。神经病变的高患病率可能被低估,因为最近几项研究报告,空腹血糖受损和/或葡萄糖耐量受损的患者在诊断时也表现出神经病变。糖尿病神经病变是西方世界足部溃疡和非创伤性截肢的最常见原因。患有糖尿病神经病变的患者报告会继发有疼痛、残疾和复发住院治疗的生活质量差等。
据估计,在美国,糖尿病神经病变的年度成本接近110亿美元,并且随着糖尿病发病率和患病率的惊人增加而逐年增加(www.diabetes.org)。
除了血糖控制和精细的足部护理之外,糖尿病神经病变没有其他治疗方法[1,3,7-9]。尽管目前正在进行研究以解决该疾病的发病机制,其目标是确定基于机制的治疗方法。近年来,已经出现了这样的想法:由于多种不同的代谢途径受损,导致共同的最终结果:不断放大的细胞内氧化应激。本综述将重点关注线粒体和细胞溶质氧化应激之间的关系以及不断放大的细胞内氧化应激和糖尿病神经病变的发生和发展的生化途径。读者可参考以下最近的综述文章,了解糖尿病神经病变的症状,分期和治疗[1-7,10]。
1.1 氧化应激:线粒体中活性氧/氮物种(ROS / RNS)的形成
老化的“自由基理论”是由Harman在1956年提出的。根据他的衰老理论,“通常在生物体中产生的活性自由基与细胞成分的反应引发了与衰老相关的变化。”过剩的自由生成自由基导致压力信号的上调,对生活质量和寿命产生负面影响。活性氧(ROS)和活性氮(RNS)物种与多种疾病状态有关[11-15],包括糖尿病的微血管并发症[16-20]。线粒体代谢和氧化磷酸化级联被强调为许多疾病中ROS产生的关键因素。
线粒体氧化磷酸化是真核生物中主要的ATP合成途径。在该过程中,来自还原底物的电子通过呼吸链复合物I-IV转移至分子氧(O2)。这些复合物在线粒体内膜上建立氢梯度,然后该梯度的电化学能量用于通过ATP合酶(复合物V)驱动ATP合成。
有四种与呼吸链相关的蛋白质复合物。 NADH-泛醌氧化还原酶或复合物I接受来自NADH的电子;这些电子被带到琥珀酸脱氢酶,复合物II,并用于将琥珀酸氧化成富马酸盐。电子继续沿电化学梯度向下移动至泛醇 - 细胞色素c氧化还原酶(复合物III),随后向细胞色素c氧化酶(复合物IV)传播,最终用于将分子氧还原为水。即使大部分分子氧在复合物IV中通过呼吸链还原为水,1-4%的氧气也不完全还原为超氧化物(O2∙ - )[21]。
O2∙ - 是最常见的ROS,通过本综述后面讨论的各种酶促或非酶促反应产生其他ROS / RNS。
O2∙ - 线粒体电子传递链的产生主要在复合物I和III上。有人认为O2∙ - 复合物Ⅰ中的生成是通过反向电子转移,主要释放到基质中[22]。
复合物III中的泛半醌自由基中间体(QH∙)的自氧化是O2∙ - 产生的另一个来源。复合物III具有向线粒体内膜两侧释放O2∙的能力,然而,更接近膜间隙(Qo)的Q位点,已知是O2∙ - 生产的主要位点,并且基质侧(Qi)不太可能形成O2∙ - [23,24]。
如上所述,O2∙ - 是线粒体间隙或基质中产生的主要ROS。作为带电荷的活性物质,O2∙ - 不易扩散穿过线粒体膜。然而,线粒体通透性转换孔可能作为膜间线粒体O2∙ - 的通道通过线粒体外膜进入细胞质[25]。通过超氧化物歧化酶(SOD)将O 2∙ - 转化为过氧化氢(H 2 O 2)促进通过产生不带电的ROS的渗透,所述不带电的ROS容易地扩散穿过膜。
有三种同种型的SOD,SOD1或铜锌SOD(CuZn-SOD),SOD2或锰SOD(Mn-SOD),SOD3或细胞外CuZn-SOD(EC-SOD)[26]。 CuZn-SOD(SOD1)存在于线粒体的细胞质,细胞核和细胞间隙[27],Mn-SOD(SOD2)仅在线粒体基质中表达,SOD3位于细胞外空间。在所有SOD同种型中,SOD2在生理学上更重要,与其他同种型相比,其遗传上的消失,会导致胚胎致死[28,29]。
过氧化氢酶和谷胱甘肽过氧化物酶酶促还原H2O2。在线粒体基质中,谷胱甘肽过氧化物酶使用谷胱甘肽将H2O2转化为水。与谷胱甘肽过氧化物酶相比,过氧化氢酶对H2O2具有更高的Km,并且可以防止更高浓度的H2O2 [30]。其他抗氧化酶如谷胱甘肽S-转移酶和硫氧还蛋白[31]也有助于去除和灭活线粒体中形成的ROS。在过渡金属如铜和铁的存在下,H2O2通过芬顿反应或Haber-Weiss反应产生羟基(OH)。羟基自由基具有很高的反应性,通过DNA和蛋白质修饰对局部细胞器损伤非常显著。
2、调节线粒体ROS产生
在氧化磷酸化过程中,电子携带的能量被配合物I,III和IV用于将质子泵出基质。 ATP合成酶使用跨线粒体内膜产生的电化学梯度来驱动ADP合成ATP。
在线粒体中,通过解偶联蛋白调节增加的ATP合成。在解偶联蛋白(UCP)激活后,质子穿过内膜并从ATP合酶中“解耦”氧化代谢,导致ATP产生的丧失。在过度表达UCP的背根神经节感觉神经元中,基础和高血糖诱导的ROS形成减少[32]。通过O2-激活UCP增加线粒体膜通透性,导致电化学电位降低和O2-代的进一步减少。温和的线粒体去极化限制了Ca2 +的积累并减少了活性物质的产生(例如通过限制一氧化氮合酶,NOS,活性)可以解释UPC的保护作用[33]。
线粒体ROS也受一氧化氮(NO)的调节,一氧化氮是由NOS产生的可扩散气体。 Ghafourifar和Richter在1997年报道了线粒体NOS(Mt NOS)及其活性的存在[34]。 MtNOS与线粒体内膜的基质面相关。 NOS的活性受线粒体内Ca2 +浓度即[Ca2 +] m [34]的调节。
[Ca2 +] m的升高增加NO的产生并导致Δψ的减少,而Δψ的减少从线粒体释放Ca2 +并导致MtNOS失活。 其他ROS / RNS在线粒体中由O2∙ - 和其他活性物质的相互作用产生。 在NO存在下产生O2.-导致过氧亚硝酸盐(ONOO-)形成。 ONOO-形成的速率为9.5×10-8M s-1,超过NO与细胞色素c氧化酶(0.8×10-8M s-1)的相互作用[35,36]。Ghafourifar等报道,过氧亚硝酸盐诱导的应激促进细胞色素c从线粒体中释放并导致细胞凋亡[37]。 Mt NOS也参与线粒体功能障碍。 蛋白质的酪氨酸残基[38-40]的硝化和蛋白质硫醇的S-亚硝化是线粒体中非常重要的反应[41]。
3、ROS/RNS和糖尿病神经病变
如上所述,在正常条件下,神经元具有中和ROS和RNS的能力[42-44]。 因为O2.-和H2O2是线粒体电子传递链的正常产物,SOD、过氧化氢酶和谷胱甘肽通常足以去除这些代谢副产物(图1)[45]。 然而,高血糖症增加线粒体活性和随后的O2-产生。 如前一节所述,这种初级线粒体ROS的过量产生导致RNS的形成。 因此,过量的线粒体活性导致神经元中ROS和RNS的压倒性产生,所述神经元已经耗尽还原当量并且与其他代谢和炎性损伤引起的氧化应激作斗争(下文综述)。ROS / RNS在神经元中的累积加上神经元无法解毒过量的ROS和RNS导致进行性细胞器、膜和核功能障碍。

【 图1 :氧化应激和线粒体功能障碍[45]。高血糖增加线粒体中活性氧(ROS)的产生。由三羧酸循环产生的NADH和FADH2转移至线粒体,在那里它们作为线粒体膜相关氧化还原酶复合物的电子供体。电子(e-)通过氧化还原酶复合物I,II,III和IV(细胞色素c)穿梭,直到它们被传递给分子氧,形成水。通过NADH(和FADH2通过复合物II至复合物III)将电子转移到复合物I,III和IV中,在线粒体外膜处产生质子梯度,在线粒体内膜和线粒体外膜之间产生电位。这种潜力驱动ATP合成,对线粒体活力、功能和正常代谢至关重要。然而,当电子从复合物II传递到复合物III时,ROS作为副产物产生。在正常氧化磷酸化过程中产生的ROS水平很小,并且它们被细胞抗氧化剂如谷胱甘肽、过氧化氢酶和超氧化物歧化酶解毒。另一方面,高血糖细胞通过糖酵解和三羧酸循环使更多葡萄糖穿梭,从而为细胞提供过量的NADH和FADH2电子供体。这在线粒体内膜上产生高质子梯度,这增加了初始复合物的周转,从而产生增加的自由基水平。这些自由基或ROS的积累对线粒体DNA,线粒体膜和整个细胞严重不利。缩写:Cyto-c细胞色素c,CoQ10辅酶Q10,电子电子,GSH谷胱甘肽,GSSG氧化型谷胱甘肽,H2O2过氧化氢,O2 - 超氧化物,磷酸盐,SOD超氧化物歧化酶】
值得注意的是,线粒体既是ROS / RNS生成的来源,也是第一个被破坏的结构,使神经元处于更大的风险之中。鉴于糖尿病神经病变的典型远端近端长度依赖性进展,轴突特别容易导致糖尿病神经病变的代谢和血管失衡[45]。轴突易患高血糖症,不仅因为它们直接获得神经血液供应,还因为它们有大量的线粒体。越来越多的证据表明,轴突即使不是更多,也容易受到ROS和RNS介导的损伤,部分原因是它们依赖局部线粒体获得能量。随着这些线粒体逐渐失调,轴突会发生能量衰竭,从而导致轴突变性[45-47]。
线粒体也是细胞存活信号传导途径的关键调节因子,并且毫不奇怪,线粒体DNA,蛋白质和膜的氧化损伤启动信号传导途径,随后导致细胞凋亡。
在明确细胞凋亡发生之前,由氧化应激损伤的线粒体通过称为细胞凋亡的局部过程被破坏。细胞凋亡的部分调节是通过改变正常线粒体裂变/融合平衡的平衡。线粒体的裂变是由发动蛋白相关蛋白1(Drp1)引发的,它在应激期间从细胞质转移到线粒体[48]。过量的线粒体裂变导致细胞凋亡,然后可能发展为细胞凋亡。 Drp1在糖尿病神经病变的体外和体内模型中升高[45],进一步促进线粒体功能障碍、能量衰竭和轴突变性。
虽然许多研究记录了糖尿病感觉神经元中轴突营养不良和细胞凋亡的存在[49],但一些研究未能检测到培养物中高糖处理的感觉神经元的细胞凋亡[50,51]。一个假设认为,神经元在周围神经胶质提供的营养因子和抗氧化剂的支持下,最初能够成功修复。然而,最终,葡萄糖介导的ROS / RNS累积的循环导致线粒体损伤和上文概述的损伤级联:能量衰竭和轴突变性[52,53]。
4、ROS / RNS与其他代谢途径的交集
过量产生线粒体ROS / RNS是糖尿病神经病变中高血糖诱导的神经损伤的关键步骤。由于持续的高血糖症,其他几种代谢途径在神经系统中受到干扰。这些途径中的每一种都有助于糖尿病性神经病中存在的神经元和轴突损伤,并且还有助于提高糖尿病神经系统中存在的氧化应激水平。在以下部分中,将讨论这些途径中的每一个,并且向读者提供一个或多个参考文献以对各个途径进行彻底的评论。图2是糖尿病性神经病变产生途径相互作用的示意图; 这些路径按照它们在图2中从左到右的顺序进行讨论。
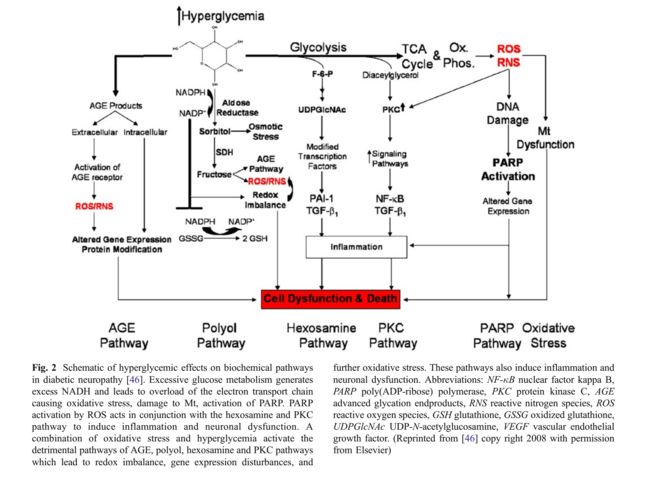
【图2:糖尿病神经病变对生化途径的高血糖作用示意图[46]。过量的葡萄糖代谢产生过量的NADH并导致电子传递链过载,引起氧化应激,损害Mt,激活PARP。 ROS的PARP活化与己糖胺和PKC途径一起起作用以诱导炎症和神经元功能障碍。氧化应激和高血糖的组合激活AGE、多元醇、己糖胺和PKC途径的有害途径,其导致氧化还原失衡,基因表达紊乱和进一步的氧化应激。这些途径还诱导炎症和神经元功能障碍。缩写:NF-κB:核因子κB;PARP:聚(ADP-核糖)聚合酶;PKC:蛋白激酶C;AGE:晚期糖基化终产物;RNS:活性氮物种;ROS:活性氧物种;GSH:谷胱甘肽;GSSG:氧化型谷胱甘肽;UDPGlcNAc: UDP-N - 乙酰氨基葡萄糖;VEGF:血管内皮生长因子。(2008年版权所有,经Elsevier许可,转载自[46]副本】
当读者回顾被认为产生糖尿病神经病变的其他代谢途径时,重要的是要记住糖尿病神经病变是由高血糖诱导的神经细胞和轴突本身的损伤以及由高血糖引起的神经血管血流减少引起的神经元缺血引起的。我们总结了最近的实验性糖尿病和神经病变动物模型以及表1中的周围神经系统氧化应激的证据。
4.1高级糖基化终产物(AGE)途径
晚期糖基化终产物(AGEs)是还原糖或氧化醛与蛋白质、DNA或脂质之间的非酶促产生的加合物[72,73]。
因此,AGEs是异质的,并且在细胞内部和外部被发现,其中它们的形成干扰细胞功能的多个方面。反应性二羰基是AGEs的前体分子,其自发地与蛋白质或脂质形成共价键,并且通过三种途径合成:葡萄糖氧化,其形成乙二醛;果糖 - 赖氨酸加合物的降解(Amadori产品);通过糖酵解中间体的异常代谢形成甲基乙二醛。
甲基乙二醛具有高度反应性,可使血管内皮细胞对损伤更敏感[74]。蛋白质AGEs的细胞外形成不仅破坏细胞粘附(通过干扰细胞表面蛋白/细胞外基质相互作用),而且还激活AGEs的特定细胞表面受体,称为RAGE [75]。
细胞外AGEs激活RAGE导致转录因子核因子κB(NF-κB)的活化,其调节基因表达、细胞凋亡和炎症(图2)。糖尿病动物模型中的RAGE活化有助于糖尿病神经病的发作和进展。当RAGE敲除小鼠用链脲佐菌素(STZ)制成糖尿病时,与STZ对照动物相比,糖尿病神经病变的电生理学和解剖学标记物都有显著改善。糖尿病RAGE敲除小鼠的外周神经中NF-κB和蛋白激酶C的表达也降低,尤其是施万细胞[73]。
神经元中的RAGE激活也会诱导NADPH氧化酶活性,进一步促进线粒体氧化应激和功能障碍[76]。数据的汇合有力地表明RAGE是治疗糖尿病神经病变的治疗靶点[73,76-81]。为了详细讨论AGE和RAGE在糖尿病神经病变的发病机制中的作用以及阻断RAGE治疗糖尿病神经病变的潜在治疗效果,读者可参考Sugimoto及其同事[82]的优秀2008年评论。
4.2 多元醇途径
多元醇途径通过两步还原/氧化将葡萄糖转化为果糖:首先,醛糖还原酶将葡萄糖还原为山梨糖醇,然后山梨糖醇脱氢酶将山梨糖醇氧化成果糖(图2)。醛糖还原酶和山梨糖醇脱氢酶在易于发生糖尿病并发症的组织中普遍存在。醛糖还原酶途径易受高血糖的质量反应作用的过度激活,这导致两种途径代谢物,NADPH和山梨糖醇的不平衡。
过量的葡萄糖流过该途径导致NADPH的消耗,这是还原型谷胱甘肽再生所需的[83,84]。如上所述,继发于过量醛糖还原酶活性的谷胱甘肽的消耗因此使细胞易受氧化应激的影响。
增加山梨醇的产生导致细胞内环境变得高渗,并导致其他渗透物如肌醇(MI,在信号转导中很重要)和牛磺酸(抗氧化剂)的代偿性流出[19,85]。通过多元醇途径的第二步,即果糖的产生,细胞内还原电位进一步降低[86]。高血糖驱动的过量果糖的产生促进了糖化并进一步消耗了NADPH。最后,醛糖还原酶的活化还可以增加二酰基甘油的形成,其激活有害的蛋白激酶C途径(下面讨论)[87,88]。人醛糖还原酶基因的几项研究揭示了与糖尿病并发症易感性相关的多态性。具有“高醛糖还原酶表达”基因型的患者通常被发现具有早期糖尿病神经病变,而具有“低醛糖还原酶表达”基因型的患者对神经病变较不敏感[89-91]。
多元醇途径已经并将继续成为糖尿病神经病变治疗中药物干预的目标。醛糖还原酶抑制剂阻止山梨糖醇的形成,防止NADPH消耗。这留下了足够的NADPH用于谷胱甘肽生成,从而允许神经元发起针对ROS / RNS介导的损伤的细胞防御。现在人们普遍认为,这种作用机制是醛糖还原酶抑制剂的有益作用的基础。 Peter Oates最近发表了对多元醇途径以及过去和目前醛糖还原酶抑制剂试验的全面综述[92]。到目前为止,这些化合物中没有一种在糖尿病神经病变的3期试验中显示出功效。
4.3己糖胺途径
与多元醇途径一样,过量的可用葡萄糖导致通过己糖胺途径的通量的质量作用增加。在正常情况下,少量的糖酵解中间体果糖-6磷酸从糖酵解分流到己糖胺途径。己糖胺途径通过谷氨酰胺果糖-6磷酸酰胺转移酶将果糖-6磷酸转化为葡糖胺-6磷酸[93]。然后将氨基葡萄糖-6磷酸转化为尿苷二磷酸N-乙酰葡糖胺(UDP-GlcNAc),它是O-GlcNAc转移酶的必需底物,将O-GlcNAC连接到转录因子的丝氨酸和苏氨酸残基并改变基因表达[16] 。
因此,通过该途径的高血糖驱动的通量增加导致基因表达的异常[16,94,95]。对O-GlcNAC生物学的更多了解也表明O-GlcNACcylation调节己糖胺途径的营养传感作用,并在胰岛素抵抗和大血管并发症中发挥作用[96,97]。
Sp1是涉及糖尿病并发症的一种转录因子,其可通过UDPGlcNAc修饰。 Sp1调节许多葡萄糖诱导的“管家”基因的表达,包括组织型纤溶酶原激活物抑制剂-1(PAI-1)和转化生长因子-β1(TGF-β1)[16,98]。对纤溶酶原激活物和PAI-1的兴趣基于这样的前提:小神经血管中纤维蛋白溶解受损促进神经缺血,导致氧化应激和糖尿病神经病变的体征和症状。支持这一想法的数据主要来自人的研究。糖尿病神经病变患者的腓肠神经和神经内膜微血管中的纤溶酶原激活物表达降低了4至6倍。这种较低的表达会促进血栓形成和神经缺血[99]。在糖尿病干预和并发症流行病学研究(EDIC)中,1型糖尿病男性的数据进一步支持了这一观点。糖尿病神经病变患者血清纤溶酶原激活物/ PAI-I复合物水平高于无神经病变患者[100]。 2型肥胖患者的PAI-I水平较高,这可能导致该人群中糖尿病神经病变的高发[101,102]。在实验动物中,PAI-I阻断神经再生[103]。糖尿病神经病变实验模型需要做更多的工作才能充分了解纤溶酶原激活物/ PAI-I复合物的作用。
TGF-β1的过度表达与糖尿病肾病有关,并通过刺激胶原基质的产生和抑制肾小球系膜细胞的有丝分裂而导致微血管损伤[104,105]。 Russell实验室最近的一项研究确定了TGF-β和其他TGF亚型在实验性糖尿病神经病变中的作用。在STZ糖尿病12周后,TGF-β同种型在具有神经病的啮齿动物的背根神经节和坐骨神经中增加。同时,TGF-β同种型直接应用于背根神经节培养物体外阻断神经突向外生长[106]。总的来说,这些新发现表明TGF-β可能是糖尿病神经病变的潜在新靶点,类似于其在糖尿病肾病中的作用。
4.4、蛋白激酶C(PKC)途径
高血糖通过增加激活PKC的二酰基甘油(DAG)的合成来刺激蛋白激酶C(PKC)途径的过度活化。特别是PKCβ-同种型与视网膜病变、肾病和心血管疾病的发展有关[107-109]。 PKC的过度刺激导致血管生成蛋白血管内皮生长因子(VEGF)、PAI-1、NF-κB和TGF-β的过表达,支持PKC活化在糖尿病性神经病变的发病机制中的作用(图2)。对STZ糖尿病大鼠的研究表明,PKC-β抑制剂可改善糖尿病神经病变的测量,包括坐骨神经血流和神经传导速度[85,110]。虽然PKC-β对糖尿病神经病变的确切机制需要进一步研究,但PKC诱导的血管收缩、毛细血管通透性改变、缺氧和神经基底膜增厚都被认为是原因[107,108]。 PKC同种型的过表达也直接诱导胰岛素抵抗,这可能进一步促成糖尿病神经病变的发作[111,112]。用PKC抑制剂ruboxistaurin治疗有症状的糖尿病神经病变患者并未导致临床改善[113,114],这可能是由于药物不能穿透血液神经屏障[108]。最近的综述完全讨论了PKC和糖尿病微血管和大血管并发症的作用以及PKC抑制剂的治疗效果[108]。
4.5 Poly-ADP核糖聚合酶(PARP)途径
PARP是一种与氧化性亚硝化应激密切相关的核酶,在感觉神经元,雪旺氏细胞和内皮细胞中表达。虽然高血糖,自由基和氧化剂刺激PARP活化,PARP也会引起氧化应激(图2)[115]。 PARP将烟酰胺腺嘌呤二核苷酸(NAD +)切割成烟酰胺,还可去除附着在核蛋白上的ADP-核糖残基[116]。 PARP的催化活性会引起许多有害影响,包括基因表达的变化,自由基和氧化剂浓度的增加,NAD +的消耗,以及糖酵解中间体向其他致病途径的分流,这些途径可导致PKC活化和AGE形成[64,117- 119]。在实验性糖尿病中,这些不同的效应导致神经血管异常、神经病变、神经传导速度降低,热痛和机械痛觉过敏以及触觉异常性疼痛[44,45,67,120-122]。最近的一些综述概述了PARP激活在糖尿病神经病变中的作用,并讨论了新出现的PARP靶向疗法[123,124]。
4.6、炎症
血液中炎症蛋白水平升高,包括C反应蛋白和TNF-α,与神经病变有关[125,126]。 Hsp27是导致炎症介质环氧合酶-2(Cox-2)、IL-6和IL-8释放的TNF-α信号通路的一部分,最近被Eurodiab研究发现在血液中升高糖尿病患者的神经病变[127]。正如本综述前面部分所讨论的,一些炎症介质如TNF-α和TGF-β受高血糖驱动的代谢和信号传导异常调节[17,20]。己糖激酶和PKC途径中过量的葡萄糖介导的活性导致信号中间体和修饰的转录因子的激活,最终增加TGF-β和NF-κB[16]。类似地,AGE甲基乙二醛的形成导致共价修饰的转录因子,其可导致炎性蛋白的异常表达,特别是称为Sp3的血管紧张素II的阻遏物[74]。由此产生的可用血管紧张素II的增加激活了血管内皮细胞[74]。神经内膜中活化的内皮细胞募集炎症细胞,导致局部细胞因子产生、血流减少和活性氧的产生[58]。
细胞外AGEs激活RAGE也会通过引起NF-κB的上调来影响炎症[73],从而上调Cox-2 [128]。 Cox-2激活导致前馈环:Cox-2刺激前列腺素E2和ROS的产生,其继续激活NF-κB。 NF-κ-B / Cox-2上调存在于糖尿病动物模型的脉管系统和外周神经中[129]。阻断Cox-2上调药物或基因敲除可以预防糖尿病神经病变的多个方面,包括血流和神经传导缺陷,谷胱甘肽耗竭和TNFα上调[65,130]。
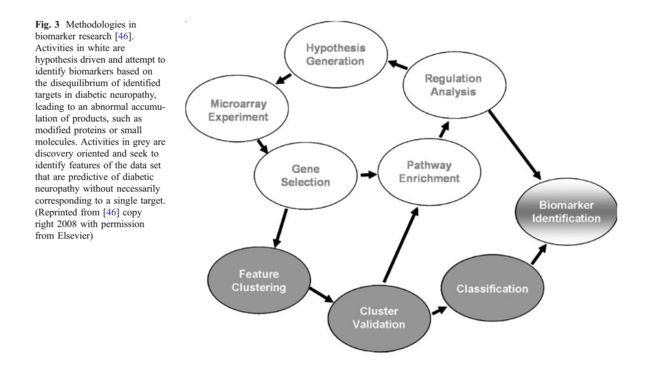
【图3生物标志物研究中的方法[46]。白色活动是假设驱动,试图基于糖尿病神经病变中识别靶部位的不平衡来鉴定生物标志物,导致产物(例如修饰的蛋白质或小分子)的异常积累。 灰色活动是基于发现和寻求识别预测糖尿病性神经病变的数据集特征,而不必对应于单个目标。 (2008年版权所有,经Elsevier许可,转载自[46]副本)】
NF-κB参与炎症的第二个恶性循环,其中它诱导并诱导一氧化氮合酶(iNOS)诱导[131,132]。由过量的iNOS产生的NO通过减少神经的血液供应而导致微血管损伤[133,134]。
此外,NO有助于损伤后的轴突和髓鞘变性,损害生长锥,并参与神经性疼痛的发展[133,135]。
NF-κB似乎是参与糖尿病神经病变发展的炎症途径的基石。慢性NF-κB活化似乎使神经元和血管更容易受到缺血再灌注损伤[136]。 NF-κB刺激内皮细胞,许旺细胞和神经元细胞因子的释放进一步加剧了随后巨噬细胞的广泛浸润[137]。巨噬细胞的活化导致细胞因子的进一步产生,以及导致髓鞘分解,细胞氧化损伤和神经再生受损的蛋白酶和ROS [138-140]。 Cameron和Cotter [141]最近回顾了NF-κB在糖尿病神经病变中的作用以及旨在减少炎症以阻止糖尿病神经病变进展的新疗法。
5、寻找新的治疗目标
葡萄糖控制仍然是糖尿病神经病变的唯一疾病改善疗法[7,18,47]。我们提出生物信息学方法作为检查糖尿病神经病变的原因和潜在治疗方法的下一个重要范例。这种新颖的范例将提供对疾病发病机理的深入了解,并确定疾病调节治疗的可行目标。对糖尿病神经病变患者和动物模型的基因组和蛋白质组学数据的分析不仅会验证或反驳当前的假设,还会产生新的想法,以进一步增强我们对疾病发作和进展的理解。
迄今为止,只有两项动物研究(并没有人类研究)已经解决了在高血糖应激和/或糖尿病的常见治疗下周围神经系统内基因表达的改变[54,142]。 Price等[142]对STZ诱导的糖尿病后第1,4和8周Wistar大鼠背根神经节神经元进行了微阵列分析。在第4周和第8周,神经传导速度减慢证实了糖尿病神经病变[142]。在神经病变发作之前诱导糖尿病与参与葡萄糖代谢的基因的上调相关[142]。到第4周,谷胱甘肽转移酶继发于氧化应激条件[142]。我们最近报道(2008),与代谢、线粒体、金属离子结合和一般细胞调节功能相关的基因在1型糖尿病和健康小鼠的坐骨神经中显着差异表达[54]。线粒体基因表达的变化与近端启动子中NF-κB和AP1结合位点的存在有关,与转录因子结合位点的整体分布相比,在受调节的线粒体基因中过量表达10倍。在小鼠启动子区域。用罗格列酮治疗糖尿病小鼠,减少神经病变的发展,减少神经中的氧化应激[54]。
通过罗格列酮处理将由糖尿病显着调节然后恢复至正常水平的基因分析常见的转录因子结合位点,并且在用罗格列酮处理的健康小鼠中交叉检查结果以确定因果关系的方向。两个站点组合,SP1F和ZBPF,以及两个EGRF站点的配置被发现两者都很重要方法[54]。我们和其他人正在全面努力建立包括外周神经和感觉神经节和交感神经节在内的神经组织的分子特征,来自人类患者的RNA和有和没有糖尿病神经病变的动物模型的全基因组筛选[52,143]。我们对糖尿病神经病变的方法如图3所示。
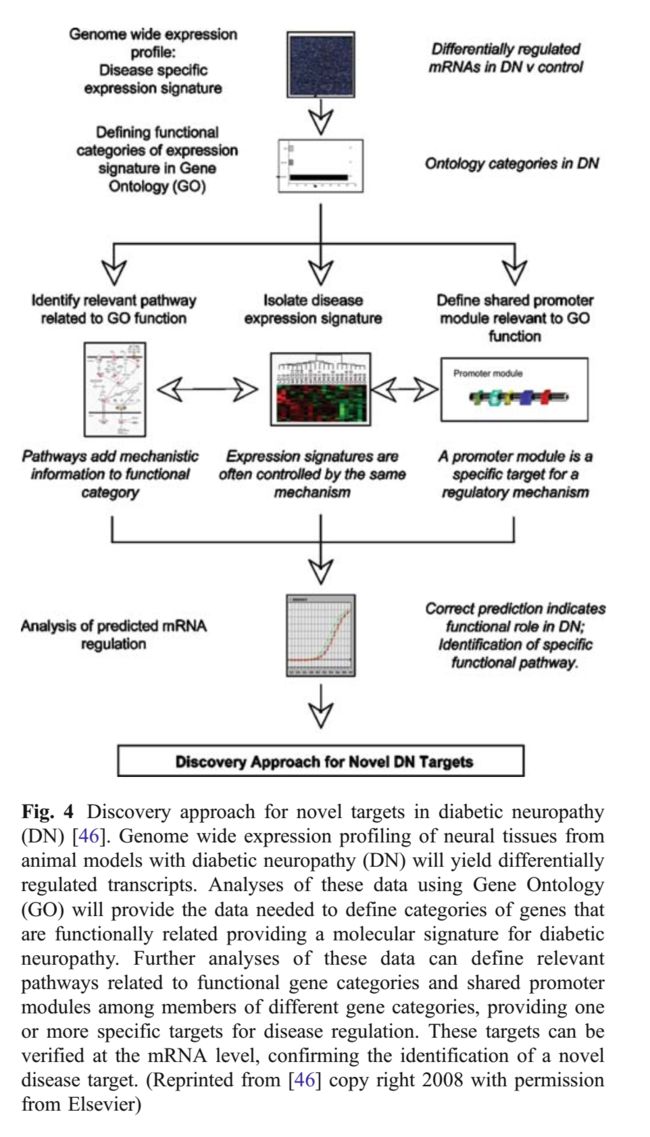
Kretzler及其同事[144-147]描述了使用发现/生物信息学方法揭示疾病机制的可行性、力量和效用。通过Affymetrix TM微阵列分析和实时RT-PCR检查的人肾活检显示健康组织和患病组织之间差异表达的基因。将这些基因定位于已知的细胞途径,预测调节元件控制观察到的变化。然后使用调节元件预测基因表达的下游效应,包括慢性肾病的潜在生物标志物。我们提出了一种类似的综合方法应用于糖尿病神经病变,以促进我们对疾病发病机制和疾病缓解疗法的发展的理解。特别感兴趣的是这种方法同时如何导致生物标志物的发现。如图4所示,第一步是采用微阵列分析和验证性Q-PCR分析相关神经元和施万细胞标记物的基因表达,然后通过验证技术检测富集途径。这些数据提供了预测受基因表达影响的蛋白质和大分子所需的信息。使用聚类和分类分析,同时保持高标准的数学验证,是发现最有用的靶基因,蛋白质和大分子的有价值的工具。
6、总结
正在进行的研究表明,多种代谢和血管通路交叉产生全身和神经氧化应激,这是糖尿病神经病变发生和发展的基础。 基于当前综述中讨论的机制的疗法尚未成功改善疾病进展。 我们建议对糖尿病神经病变采用新的生物信息学方法,为将来鉴定更有希望的分子靶标提供前景。
致谢我们感谢Judith Boldt女士的出色秘书支持以及Kelli A. Sullivan博士的专家编辑建议。 这项工作得到了A. Alfred Taubman医学研究所,神经病学研究和发现计划以及NIH T32 NS07222和NIH UO1 DK076160的支持。
References
1. Boulton AJ, Vinik AI, Arezzo JC, Bril V, Feldman EL, Freeman R, et al. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care 2005;28:956–62. doi: 10.2337/diacare.28.4.956.
2. Martin CL, Albers J, Herman WH, Cleary P, Waberski B, Greene DA, et al. Neuropathy among the diabetes control and complications trial cohort 8 years after trial completion. Diabetes Care 2006;29:340–4. doi:10.2337/diacare.29.02.06.dc05-1549.
3. Feldman EL, Stevens MJ, Russell JW, Peltier A, et al. Somatosensory neuropathy. In: Porte D Jr, Sherwin RS, Baron A, editors. The diabetes mellitus manual. Philadelphia: McGrawHill; 2005. p. 366–84.
4. Singleton JR, Smith AG, Russell J, Feldman EL. Polyneuropathy with impaired glucose tolerance: implications for diagnosis and therapy. Curr Treat Options Neurol 2005;7:33–42. doi:10.1007/s11940-005-0004-4
5. Feldman EL, Stevens MJ, Russell JW, Greene DA, Porte D Jr,Sherwin RS, et al. Somatosensory neuropathy. In: Porte D Jr SRS, Baron A, editors. Ellenberg and Rifkin’s diabetes mellitus. Philadelphia: McGraw Hill; 2002. p. 771–88.
6. Feldman EL, Stevens MJ, Russell JW. Diabetic peripheral and autonomic neuropathy. In: Sperling MA, editor. Contemporary endocrinology. Humana Press; 2002.
7. Little AA, Edwards JL, Feldman EL. Diabetic neuropathies. Pract Neurol 2007;7:82–92.
8. Vinik AI, Maser RE, Mitchell BD, Freeman R. Diabetic autonomic neuropathy. Diabetes Care 2003;26:1553–79. doi:10.2337/diacare.26.5.1553.
9. Vinik AI, Mehrabyan A. Diabetic neuropathies. Med Clin North Am 2004;88:947–99. doi:10.1016/j.mcna.2004.04.009xi.
10. Feldman EL, Stevens MJ, Thomas PK, Brown MB, Canal N, Greene DA. A practical two-step quantitative clinical and electrophysiological assessment for the diagnosis and staging of diabetic neuropathy. Diabetes Care 1994;17:1281–9. doi:10.2337/diacare.17.11.1281.
11. Hoye AT, Davoren JE, Wipf P, Fink MP, Kagan VE. Targeting mitochondria. Acc Chem Res 2008;41:87–97. doi:10.1021/ ar700135m.
12. Lee MY, Griendling KK. Redox signaling, vascular function, and hypertension. Antioxidants & Redox Signaling 2008;10:1045–59.
13. Lu T, Finkel T. Free radicals and senescence. Exp Cell Res 2008;314:1918–22. doi:10.1016/j.yexcr.2008.01.011.
14. Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care 2008;31 (Suppl 2):S170–80. doi:10.2337/dc08-s247
.15. Skulachev VP. A biochemical approach to the problem of aging: “megaproject” on membrane-penetrating ions. The first results and prospects. Biochemistry 2007;72:1385–96.
16. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001;414:813–20. doi:10.1038/414813a
17. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes 2005;54:1615–25. doi:10.2337/diabetes.54.6.1615
18. Vincent AM, Edwards JL, Sadidi M, Feldman EL. The antioxidant response as a drug target in diabetic neuropathy. Curr Drug Targ ets 2008;9:94–100. doi:10.2174/138945008783431754
19. Vincent AM, Feldman EL. New insights into the mechanisms of diabetic neuropathy. Rev Endocr Metab Disord 2004;5:227–36. doi:10.1023/B:REMD.0000032411.11422.e0.
20. Vincent AM, Russell JW, Low P, Feldman EL. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr Rev 2004; 25:612–28. doi:10.1210/er.2003-0019.
21. Erusalimsky JD, Moncada S. Nitric oxide and mitochondrial signaling: from physiology to pathophysiology. Arterioscler Thromb Vasc Biol 2007;27:2524–31. doi:10.1161/ATVBAHA.107.151167.
22. Lambert AJ, Brand MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH: ubiquinone oxidoreductase (complex I). J Biol Chem 2004;279:39414–20. doi:10.1074/jbc.M406576200.
23. Han D, Williams E, Cadenas E. Mitochondrial respiratory chaindependent generation of superoxide anion and its release into the intermembrane space. Biochem J 2001;353:411–6. doi:10.1042/ 0264-6021:3530411
24. Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem 2004;279:49064–73. doi:10.1074/jbc.M407715200
25. Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltagedependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem 2003;278:5557–63. doi:10.1074/jbc.M210269200.
26. Faraci FM, Didion SP. Vascular protection: superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vasc Biol 2004;24:1367–73. doi:10.1161/01.ATV.0000133604.20182.cf.
27. Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver. Cu,Zn-SOD in mitochondria. J Biol Chem 2001;276:38388–93. doi:10.1074/jbc.M105395200
28. Lebovitz RM, Zhang H, Vogel H, Cartwright J Jr, Dionne L, Lu N, et al. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA 1996;93:9782–7. doi:10.1073/pnas.93.18.9782
29. Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 1995;11:376–81. doi:10.1038/ng1295-376.
30. Suttorp N, Toepfer W, Roka L. Antioxidant defense mechanisms of endothelial cells: glutathione redox cycle versus catalase. Am J Physiol Cell Physiol 1986;251:C671–80.
31. Zhang R, Al-Lamki R, Bai L, Streb JW, Miano JM, Bradley J, et al. Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ Res 2004; 94:1483–91. doi:10.1161/01.RES.0000130525.37646.a7.
32. Vincent AM, Gong C, Brownlee M, Russell JW. Glucose induced neuronal programmed cell death is regulated by manganese superoxide dismutase and uncoupling protein-1. Endocrine Society Abstracts 2001;P1–289:210.
33. Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science (New York, NY) 2005;307:384–7.
34. Ghafourifar P, Richter C. Nitric oxide synthase activity in mitochondria. FEBS Lett 1997;418:291 –6. doi:10.1016/S0014-5793(97)01397-5.
35. Poderoso JJ, Carreras MC, Schopfer F, Lisdero CL, Riobo NA, Giulivi C, et al. The reaction of nitric oxide with ubiquinol: kinetic properties and biological significance. Free Radic Biol Med 1999;26:925–35. doi:10.1016/S0891-5849(98)00277-9
36. Poderoso JJ, Lisdero C, Schopfer F, Riobo N, Carreras MC, Cadenas E, et al. The regulation of mitochondrial oxygen uptake by redox reactions involving nitric oxide and ubiquinol. J Biol Chem 1999;274:37709–16. doi:10.1074/jbc.274.53.37709.
37. Ghafourifar P, Schenk U, Klein SD, Richter C. Mitochondrial nitric–oxide synthase stimulation causes cytochrome c release from isolated mitochondria. Evidence for intramitochondrial peroxynitrite formation. J Biol Chem 1999;274:31185–8. doi: 10.1074/jbc.274.44.31185.
38. Marcondes S, Turko IV, Murad F. Nitration of succinyl-CoA:3-oxoacid CoA-transferase in rats after endotoxin administration. Proc Natl Acad Sci USA 2001;98:7146–51. doi:10.1073/pnas.141222598
39. Aulak KS, Koeck T, Crabb JW, Stuehr DJ. Proteomic method for identification of tyrosine-nitrated proteins. Meth Mol Biol (Clifton, NJ) 2004;279:151–65.
40. Turko IV, Marcondes S, Murad F. Diabetes-associated nitration of tyrosine and inactivation of succinyl-CoA:3-oxoacid CoAtransferase. Am J Physiol Heart Circ Physiol 2001;281:H2289–94.
41. Mannick JB, Schonhoff C, Papeta N, Ghafourifar P, Szibor M, Fang K, et al. S-Nitrosylation of mitochondrial caspases. J Cell Biol 2001;154:1111–6. doi:10.1083/jcb.200104008.
42. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000;404:787–90. doi:10.1038/35008121.
43. Obrosova IG, Drel VR, Oltman CL, Mashtalir N, Tibrewala J, Groves JT, et al. Role of nitrosative stress in early neuropathy and vascular dysfunction in streptozotocin-diabetic rats. Am J Physiol 2007;293:E1645–55. doi:10.1152/ajpcell.00165.2007.
44. Obrosova IG, Drel VR, Pacher P, Ilnytska O, Wang ZQ, Stevens MJ, et al. Oxidative-nitrosative stress and poly(ADP-ribose) polymerase (PARP) activation in experimental diabetic neuropathy: the relation is revisited. Diabetes 2005;54:3435–41. doi:10.2337/diabetes.54.12.3435.
45. Leinninger GM, Edwards JL, Lipshaw MJ, Feldman EL. Mechanisms of disease: mitochondria as new therapeutic targets in diabetic neuropathy. Nat Clin Prac 2006;2:620–8.
46. Edwards JL, Vincent AM, Cheng HL, Feldman EL. Diabetic neuropathy: Mechanisms to management. Pharmacol Ther. 2008 (in press).
47. Leinninger GM, Backus C, Sastry AM, Yi YB, Wang CW, Feldman EL. Mitochondria in DRG neurons undergo hyperglycemic mediated injury through Bim, Bax and the fission protein Drp1. Neurobiol Dis 2006;23:11–22. doi:10.1016/j.nbd.2006.01.017.
48. Arnoult D, Rismanchi N, Grodet A, Roberts RG, Seeburg DP, Estaquier J, et al. Bax/Bak-dependent release of DDP/TIMM8 promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr Biol 2005;15:2112–8. doi:10.1016/j.cub.2005.10.041.
49. Brussee V, Guo G, Dong Y, Cheng C, Martinez JA, Smith D, et al. Distal degenerative sensory neuropathy in a long-term type 2 diabetes rat model. Diabetes 2008;57:1664–73. doi:10.2337/db07-1737.
50. Cheng C, Zochodne DW. Sensory neurons with activated caspase-3 survive long-term experimental diabetes. Diabetes 2003;52:2363–71. doi:10.2337/diabetes.52.9.2363.
51. Gumy LF, Bampton ET, Tolkovsky AM. Hyperglycaemia inhibits Schwann cell proliferation and migration and restricts regeneration of axons and Schwann cells from adult murine DRG. Mol Cell Neurosci 2008;37:298–311. doi:10.1016/j.mcn.2007.10.004
.52. Sullivan KA, Hayes JM, Wiggin TD, Backus C, Su Oh S, Lentz SI, et al. Mouse models of diabetic neuropathy. Neurobiol Dis 2007;28:276–85. doi:10.1016/j.nbd.2007.07.022.
53. Vincent AM, Russell JW, Sullivan KA, Backus C, Hayes JM, McLean LL, et al. SOD2 protects neurons from injury in cell culture and animal models of diabetic neuropathy. Exp Neurol 2007;208:216–27.
54. Wiggin TD, Kretzler M, Pennathur S, Sullivan KA, Brosius FC, Feldman EL. Rosiglitazone treatment reduces diabetic neuropathy in STZ Treated DBA/2J Mice. Endocrinology. 2008 (in press)
55. Gardiner NJ, Wang Z, Luke C, Gott A, Price SA, Fernyhough P.
Expression of hexokinase isoforms in the dorsal root ganglion of
the adult rat and effect of experimental diabetes. Brain Res
2007;1175:143
–54. doi:10.1016/j.brainres.2007.08.015.
56. Saini AK, Kumar HSA, Sharma SS. Preventive and curative
effect of edaravone on nerve functions and oxidative stress in
experimental diabetic neuropathy. Eur J Pharmacol
2007;568:164
–72. doi:10.1016/j.ejphar.2007.04.016.
57. Kumar A, Kaundal RK, Iyer S, Sharma SS. Effects of resveratrol
on nerve functions, oxidative stress and DNA fragmentation in
experimental diabetic neuropathy. Life Sci 2007;80:1236
–44.
doi:
10.1016/j.lfs.2006.12.036.
58. Coppey LJ, Davidson EP, Rinehart TW, Gellett JS, Oltman CL,
Lund DD, et al. ACE inhibitor or angiotensin II receptor
antagonist attenuates diabetic neuropathy in streptozotocininduced diabetic rats. Diabetes 2006;55:341
–8. doi:10.2337/
diabetes.55.02.06.db05-0885
.
59. Ilnytska O, Lyzogubov VV, Stevens MJ, Drel VR, Mashtalir N,
Pacher P, et al. Poly(ADP-ribose) polymerase inhibition alleviates experimental diabetic sensory neuropathy. Diabetes
2006;55: 1686
–94. doi:10.2337/db06-0067.
60. Kuzumoto Y, Kusunoki S, Kato N, Kihara M, Low PA. Effect of
the aldose reductase inhibitor fidarestat on experimental diabetic
neuropathy in the rat. Diabetologia 2006;49:3085
–93.
doi:
10.1007/s00125-006-0400-7.
61. Sayyed SG, Kumar A, Sharma SS. Effects of U83836E on nerve
functions, hyperalgesia and oxidative stress in experimental
diabetic neuropathy. Life Sci 2006;79:777
–83. doi:10.1016/j.
lfs.2006.02.033
.
62. Sharma SS, Sayyed SG. Effects of trolox on nerve dysfunction,
thermal hyperalgesia and oxidative stress in experimental
diabetic neuropathy. Clin Exp Pharmacol Physiol
2006;33:1022
–8. doi:10.1111/j.1440-1681.2006.04481.x.
63. Schmeichel AM, Schmelzer JD, Low PA. Oxidative injury and
apoptosis of dorsal root ganglion neurons in chronic experimental diabetic neuropathy. Diabetes 2003;52:165
–71. doi:10.2337/
diabetes.52.1.165
.
64. Obrosova IG, Mabley JG, Zsengeller Z, Charniauskaya T,
Abatan OI, Groves JT, et al. Role for nitrosative stress in
diabetic neuropathy: evidence from studies with a peroxynitrite
decomposition catalyst. FASEB J 2005;19:401
–3.
65. Kellogg AP, Wiggin TD, Larkin DD, Hayes JM, Stevens MJ,
Pop-Busui R. Protective effects of cyclooxygenase-2 gene
inactivation against peripheral nerve dysfunction and intraepidermal nerve fiber loss in experimental diabetes. Diabetes
2007;56:2997
–3005. doi:10.2337/db07-0740.
66. Ho EC, Lam KS, Chen YS, Yip JC, Arvindakshan M, Yamagishi
S, et al. Aldose reductase-deficient mice are protected from
delayed motor nerve conduction velocity, increased c-Jun NH2-
terminal kinase activation, depletion of reduced glutathione,
increased superoxide accumulation, and DNA damage. Diabetes
2006;55:1946
–53. doi:10.2337/db05-1497.
67. Obrosova IG, Li F, Abatan OI, Forsell MA, Komjati K, Pacher P,
et al. Role of poly(ADP-ribose) polymerase activation in diabetic
neuropathy. Diabetes 2004;53:711
–20. doi:10.2337/diabetes.
53.3.711
.
68. Oltman CL, Coppey LJ, Gellett JS, Davidson EP, Lund DD,
Yorek MA. Progression of vascular and neural dysfunction in
sciatic nerves of Zucker diabetic fatty and Zucker rats. Am J
Physiol 2005;289:E113
–22. doi:10.1152/ajpcell.00040.2005.
69. Vareniuk I, Pavlov IA, Drel VR, Lyzogubov VV, Ilnytska O, Bell
SR, et al. Nitrosative stress and peripheral diabetic neuropathy in
leptin-deficient (ob/ob) mice. Exp Neurol 2007;205:425
–36.
doi:
10.1016/j.expneurol.2007.03.019.
70. Drel VR, Mashtalir N, Ilnytska O, Shin J, Li F, Lyzogubov VV,
et al. The leptin-deficient (ob/ob) mouse: a new animal model of
peripheral neuropathy of type 2 diabetes and obesity. Diabetes
2006;55:3335
–43. doi:10.2337/db06-0885.
71. Obrosova IG, Ilnytska O, Lyzogubov VV, Pavlov IA, Mashtalir N,
Nadler JL, et al. High-fat diet induced neuropathy of pre-diabetes
and obesity: effects of
“healthy” diet and aldose reductase
inhibition. Diabetes 2007;56:2598
–608. doi:10.2337/db06-1176.
72. Ahmed N. Advanced glycation endproducts
—role in pathology
of diabetic complications. Diabetes Res Clin Pract 2005;67:3
–
21. doi:10.1016/j.diabres.2004.09.004.
73. Toth C, Rong LL, Yang C, Martinez J, Song F, Ramji N, et al.
Receptor for advanced glycation end products (RAGEs) and
experimental diabetic neuropathy. Diabetes. 2008;57(4):1002
–
17, Apr.
74. Yao D, Taguchi T, Matsumura T, Pestell R, Edelstein D,
Giardino I, et al. High glucose increases angiopoietin-2
transcription in microvascular endothelial cells through methylglyoxal modification of mSin3A. J Biol Chem 2007;282:31038
–
45. doi:10.1074/jbc.M704703200.
75. Ramasamy R, Yan SF, Schmidt AM. Arguing for the motion:
yes, RAGE is a receptor for advanced glycation endproducts. Mol
Nutr Food Res 2007;51:1111
–5. doi:10.1002/mnfr.200700008.
76. Vincent AM, Perrone L, Sullivan KA, Backus C, Sastry AM,
Lastoskie C, et al. Receptor for advanced glycation end products
activation injures primary sensory neurons via oxidative stress.
Endocrinology 2007;148:548
–58. doi:10.1210/en.2006-0073.
77. Ramasamy R, Vannucci SJ, Yan SS, Herold K, Yan SF, Schmidt
AM. Advanced glycation end products and RAGE: a common
thread in aging, diabetes, neurodegeneration, and inflammation.
Glycobiology 2005;15:16R
–28R. doi:10.1093/glycob/cwi053.
78. Tanji N, Markowitz GS, Fu C, Kislinger T, Taguchi A,
Pischetsrieder M, et al. Expression of advanced glycation end
products and their cellular receptor RAGE in diabetic nephropathy
and nondiabetic renal disease. J Am Soc Nephrol 2000;11:1656
–
66.
79. Coughlan MT, Forbes JM, Cooper ME. Role of the AGE
crosslink breaker, alagebrium, as a renoprotective agent in
diabetes. Kidney Int 2007;72:S54
–60. doi:10.1038/sj.
ki.5002387
.
80. Hudson BI, Schmidt AM. RAGE: a novel target for drug
intervention in diabetic vascular disease. Pharm Res
2004;21:1079
–86. doi:10.1023/B:PHAM.0000032992.75423.9b.
81. Bucciarelli LG, Wendt T, Qu W, Lu Y, Lalla E, Rong LL, et al.
RAGE blockade stabilizes established atherosclerosis in diabetic
apolipoprotein E-null mice. Circulation 2002;106:2827
–35.
doi:
10.1161/01.CIR.0000039325.03698.36.
82. Sugimoto K, Yasujima M, Yagihashi S. Role of advanced
glycation end products in diabetic neuropathy. Curr Pharm Des
2008;14:953
–61. doi:10.2174/138161208784139774.
83. Djordjevic VB. Free radicals in cell biology. Int Rev Cytol
2004;237:57
–89. doi:10.1016/S0074-7696(04)37002-6.
84. Mathers J, Fraser JA, McMahon M, Saunders RD, Hayes JD,
McLellan LI. Antioxidant and cytoprotective responses to redox
stress. Biochem Soc Symp 2004;71:157
–76.
85. Nakamura J, Kato K, Hamada Y, Nakayama M, Chaya S,
Nakashima E, et al. A protein kinase C-beta-selective inhibitor
ameliorates neural dysfunction in streptozotocin-induced diabetic
rats. Diabetes 1999;48:2090
–5. doi:10.2337/diabetes.48.10.2090.
86. Feldman EL, Stevens MJ, Greene DA. Pathogenesis of diabetic
neuropathy. Clin Neurosci (New York, NY) 1997;4:365
–70.
87. Uehara K, Yamagishi S, Otsuki S, Chin S, Yagihashi S. Effects
of polyol pathway hyperactivity on protein kinase C activity,
nociceptive peptide expression, and neuronal structure in dorsal
root ganglia in diabetic mice. Diabetes 2004;53:3239
–47.
doi:
10.2337/diabetes.53.12.3239.
88. Yamagishi S, Uehara K, Otsuki S, Yagihashi S. Differential
influence of increased polyol pathway on protein kinase C expressions between endoneurial and epineurial tissues in
diabetic mice. J Neurochem 2003;87:497
–507. doi:10.1046/
j.1471-4159.2003.02011.x
.
89. Demaine AG. Polymorphisms of the aldose reductase gene and
susceptibility to diabetic microvascular complications. Curr Med
Chem 2003;10:1389
–98. doi:10.2174/0929867033457359.
90. Donaghue KC, Margan SH, Chan AK, Holloway B, Silink M,
Rangel T, et al. The association of aldose reductase gene
(AKR1B1) polymorphisms with diabetic neuropathy in adolescents. Diabet Med 2005;22:1315
–20. doi:10.1111/j.1464-
5491.2005.01631.x
.
91. Thamotharampillai K, Chan AK, Bennetts B, Craig ME,
Cusumano J, Silink M, et al. Decline in neurophysiological
function after 7 years in an adolescent diabetic cohort and the
role of aldose reductase gene polymorphisms. Diabetes Care
2006;29:2053
–7. doi:10.2337/dc06-0678.
92. Oates PJ. Aldose reductase, still a compelling target for diabetic
neuropathy. Curr Drug Targets 2008;9:14
–36.
93. Thornalley PJ. The potential role of thiamine (vitamin B(1)) in
diabetic complications. Curr Diabetes Rev 2005;1:287
–98.
doi:
10.2174/157339905774574383.
94. Kolm-Litty V, Sauer U, Nerlich A, Lehmann R, Schleicher ED.
High glucose-induced transforming growth factor beta1 production is mediated by the hexosamine pathway in porcine
glomerular mesangial cells. J Clin Invest 1998;101:160
–9.
doi:
10.1172/JCI119875.
95. Sayeski PP, Kudlow JE. Glucose metabolism to glucosamine is
necessary for glucose stimulation of transforming growth factoralpha gene transcription. J Biol Chem 1996;271:15237
–43.
doi:
10.1074/jbc.271.25.15237.
96. Dias WB, Hart GW. O-GlcNAc modification in diabetes and
Alzheimer
’s disease. Mol Biosyst 2007;3:766–72. doi:10.1039/
b704905f
.
97. Love DC, Hanover JA. The hexosamine signaling pathway:
deciphering the
“O-GlcNAc code”. Sci STKE 2005;2005:re13.
doi:
10.1126/stke.3122005re13.
98. Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh
F, et al. Hyperglycemia-induced mitochondrial superoxide
overproduction activates the hexosamine pathway and induces
plasminogen activator inhibitor-1 expression by increasing Sp1
glycosylation. Proc Natl Acad Sci USA 2000;97:12222
–6.
doi:
10.1073/pnas.97.22.12222.
99. Hafer-Macko CE, Ivey FM, Sorkin JD, Macko RF. Microvascular tissue plasminogen activator is reduced in diabetic
neuropathy. Neurology 2007;69:268
–74. doi:10.1212/01.
wnl.0000266391.20707.83
.
100. Maser RE, Ellis D, Erbey JR, Orchard TJ. Do tissue plasminogen
activator-plasminogen activator inhibitor-1 complexes relate to
the complications of insulin-dependent diabetes mellitus?
Pittsburgh Epidemiology of Diabetes Complications Study. J
Diabetes Complications 1997;11:243
–9. doi:10.1016/S1056-
8727(96)00040-2
.
101. Aso Y, Matsumoto S, Fujiwara Y, Tayama K, Inukai T,
Takemura Y. Impaired fibrinolytic compensation for hypercoagulability in obese patients with type 2 diabetes: association
with increased plasminogen activator inhibitor-1. Metabolism
2002;51:471
–6. doi:10.1053/meta.2002.31334.
102. Erem C, Hacihasanoglu A, Celik S, Ovali E, Ersoz HO, Ukinc
K, et al. Coagulation and fibrinolysis parameters in type 2
diabetic patients with and without diabetic vascular complications. Med Princ Pract 2005;14:22
–30. doi:10.1159/000081919.
103. Nilsson A, Moller K, Dahlin L, Lundborg G, Kanje M. Early
changes in gene expression in the dorsal root ganglia after
transection of the sciatic nerve; effects of amphiregulin and PAI-
1 on regeneration. Brain Res 2005;136:65
–74. doi:10.1016/j.
molbrainres.2005.01.008
.
104. Kanwar YS, Wada J, Sun L, Xie P, Wallner EI, Chen S, et al.
Diabetic nephropathy: mechanisms of renal disease progression.
Exp Biol Med (Maywood, NJ) 2008;233:4
–11.
105. Zhu Y, Usui HK, Sharma K. Regulation of transforming growth
factor beta in diabetic nephropathy: implications for treatment.
Semin Nephrol 2007;27:153
–60. doi:10.1016/j.semnephrol.
2007.01.008
.
106. Anjaneyulu M, Berent-Spillson A, Inoue T, Choi J, Cherian K,
Russell JW. Transforming growth factor-beta induces cellular
injury in experimental diabetic neuropathy. Exp Neurol
2008;211:469
–79. doi:10.1016/j.expneurol.2008.02.011.
107. Arikawa E, Ma RC, Isshiki K, Luptak I, He Z, Yasuda Y, et al.
Effects of insulin replacements, inhibitors of angiotensin, and
PKCbeta
’s actions to normalize cardiac gene expression and fuel
metabolism in diabetic rats. Diabetes 2007;56:1410
–20.
doi:
10.2337/db06-0655.
108. Das Evcimen N, King GL. The role of protein kinase C
activation and the vascular complications of diabetes. Pharmacol
Res 2007;55:498
–510. doi:10.1016/j.phrs.2007.04.016.
109. Veves A, King GL. Can VEGF reverse diabetic neuropathy in
human subjects? J Clin Invest 2001;107:1215
–8. doi:10.1172/
JCI13038
.
110. Cameron NE, Cotter MA. Effects of protein kinase Cbeta
inhibition on neurovascular dysfunction in diabetic rats: interaction with oxidative stress and essential fatty acid dysmetabolism. Diabetes Metab Res Rev 2002;18:315
–23. doi:10.1002/
dmrr.307
.
111. Cortright RN, Azevedo JL Jr, Zhou Q, Sinha M, Pories WJ, Itani
SI, et al. Protein kinase C modulates insulin action in human
skeletal muscle. Am J Physiol 2000;278:E553
–62.
112. Naruse K, Rask-Madsen C, Takahara N, Ha SW, Suzuma K,
Way KJ, et al. Activation of vascular protein kinase C-beta
inhibits Akt-dependent endothelial nitric oxide synthase function
in obesity-associated insulin resistance. Diabetes 2006;55:691
–8.
doi:
10.2337/diabetes.55.03.06.db05-0771.
113. Casellini CM, Barlow PM, Rice AL, Casey M, Simmons K,
Pittenger G, et al. A 6-month, randomized, double-masked,
placebo-controlled study evaluating the effects of the protein
kinase C-beta inhibitor ruboxistaurin on skin microvascular
blood flow and other measures of diabetic peripheral neuropathy.
Diabetes Care 2007;30:896
–902. doi:10.2337/dc06-1699.
114. Vinik AI, Bril V, Kempler P, Litchy WJ, Tesfaye S, Price KL, et
al. Treatment of symptomatic diabetic peripheral neuropathy
with the protein kinase C beta-inhibitor ruboxistaurin mesylate
during a 1-year, randomized, placebo-controlled, double-blind
clinical trial. Clin Ther 2005;27:1164
–80. doi:10.1016/j.clinthera.
2005.08.001
.
115. Obrosova IG, Julius UA. Role for poly(ADP-ribose) polymerase activation in diabetic nephropathy, neuropathy and
retinopathy. Curr Vasc Pharmacol 2005;3:267
–83. doi:10.2174/
1570161054368634
.
116. Southan GJ, Szabo C. Poly(ADP-ribose) polymerase inhibitors.
Curr Med Chem 2003;10:321
–40.
117. Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z,
Szabo C, et al. Inhibition of GAPDH activity by poly(ADPribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest 2003;112:
1049
–57.
118. Garcia Soriano F, Virag L, Jagtap P, Szabo E, Mabley JG,
Liaudet L, et al. Diabetic endothelial dysfunction: the role of
poly(ADP-ribose) polymerase activation. Nat Med 2001;7:108
–
13. doi:10.1038/83241.
119. Apfel SC. Nerve growth factor for the treatment of diabetic
neuropathy: what went wrong, what went right, and what does
the future hold? Int Rev Neurobiol 2002;50:393
–413.
doi:
10.1016/S0074-7742(02)50083-0.
120. Li F, Drel VR, Szabo C, Stevens MJ, Obrosova IG. Low-dose
poly(ADP-ribose) polymerase inhibitor-containing combination
therapies reverse early peripheral diabetic neuropathy. Diabetes
2005;54:1514
–22. doi:10.2337/diabetes.54.5.1514.
121. Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabo E, Szabo C.
The role of poly(ADP-ribose) polymerase activation in the
development of myocardial and endothelial dysfunction in
diabetes. Diabetes 2002;51:514
–21. doi:10.2337/diabetes.51.2.514.
122. Zheng L, Szabo C, Kern TS. Poly(ADP-ribose) polymerase is
involved in the development of diabetic retinopathy via
regulation of nuclear factor-kappaB. Diabetes 2004;53:2960
–7.
doi:
10.2337/diabetes.53.11.2960.
123. Pacher P. Poly(ADP-ribose) polymerase inhibition as a novel
therapeutic approach against intraepidermal nerve fiber loss and
neuropathic pain associated with advanced diabetic neuropathy:
a commentary on
“PARP Inhibition or gene deficiency counteracts intraepidermal nerve fiber loss and neuropathic pain in
advanced diabetic neuropathy
”. Free Radic Biol Med
2008;44:969
–71. doi:10.1016/j.freeradbiomed.2007.12.020.
124. Pacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose)
polymerase pathway in human disease. Am J Pathol 2008;173:2
–
13. doi:10.2353/ajpath.2008.080019.
125. Gonzalez-Clemente JM, Mauricio D, Richart C, Broch M,
Caixas A, Megia A, et al. Diabetic neuropathy is associated
with activation of the TNF-alpha system in subjects with type 1
diabetes mellitus. Horumon To Rinsho 2005;63:525
–9.
126. Gomes MB, Piccirillo LJ, Nogueira VG, Matos HJ. Acute-phase
proteins among patients with type 1 diabetes. Diabetes Metab
2003;29:405
–11. doi:10.1016/S1262-3636(07)70051-4.
127. Gruden G, Bruno G, Chaturvedi N, Burt D, Schalkwijk C,
Pinach S, et al. Serum heat shock protein 27 and diabetes
complications in the EURODIAB prospective complications
study: a novel circulating marker for diabetic neuropathy.
Diabetes. 2008;57(7):1966
–70, Jul.
128. Lee KM, Kang BS, Lee HL, Son SJ, Hwang SH, Kim DS, et al.
Spinal NF-kB activation induces COX-2 upregulation and
contributes to inflammatory pain hypersensitivity. Eur J Neurosci
2004;19:3375
–81. doi:10.1111/j.0953-816X.2004.03441.x.
129. Kellogg AP, Pop-Busui R. Peripheral nerve dysfunction in
experimental diabetes is mediated by cyclooxygenase-2 and
oxidative stress. Antioxidants & Redox Signaling 2005;7:1521
–9.
130. Matsunaga A, Kawamoto M, Shiraishi S, Yasuda T, Kajiyama S,
Kurita S, et al. Intrathecally administered COX-2 but not COX-1
or COX-3 inhibitors attenuate streptozotocin-induced mechanical
hyperalgesia in rats. Eur J Pharmacol 2007;554:12
–7.
doi:
10.1016/j.ejphar.2006.09.072.
131. Hasnis E, Bar-Shai M, Burbea Z, Reznick AZ. Mechanisms
underlying cigarette smoke-induced NF-kappaB activation in
human lymphocytes: the role of reactive nitrogen species. J
Physiol Pharmacol 2007;58 Suppl 5:275
–87.
132. Kim YW, Zhao RJ, Park SJ, Lee JR, Cho IJ, Yang CH, Kim SG,
Kim SC. Anti-inflammatory effects of liquiritigenin as a
consequence of the inhibition of NF-kappaB-dependent iNOS
and proinflammatory cytokines production. Br J Pharmacol.
2008;154(1):165
–73, May.
133. Levy D, Zochodne DW. NO pain: potential roles of nitric oxide
in neuropathic pain. Pain Pract 2004;4:11
–8. doi:10.1111/j.1533-
2500.2004.04002.x
.
134. Zochodne DW, Levy D. Nitric oxide in damage, disease and
repair of the peripheral nervous system. Cell Mol Biol (Noisy-leGrand, France) 2005;51:255
–67.
135. McDonald DS, Cheng C, Martinez JA, Zochodne DW.
Regenerative arrest of inflamed peripheral nerves: role of nitric
oxide. Neuroreport 2007;18:1635
–40.
136. Wang Y, Schmeichel AM, Iida H, Schmelzer JD, Low PA.
Enhanced inflammatory response via activation of NF-kappaB in
acute experimental diabetic neuropathy subjected to ischemia
–
reperfusion injury. J Neurol Sci 2006;247:47–52. doi:10.1016/j.
jns.2006.03.011
.
137. Yamagishi S, Ogasawara S, Mizukami H, Yajima N, Wada R,
Sugawara A, et al. Correction of protein kinase C activity and
macrophage migration in peripheral nerve by pioglitazone,
peroxisome proliferator activated-gamma-ligand, in insulindeficient diabetic rats. J Neurochem 2008;104:491
–9.
138. Kawamura N, Dyck PJ, Schmeichel AM, Engelstad JK, Low PA,
Dyck PJ. Inflammatory mediators in diabetic and non-diabetic
lumbosacral radiculoplexus neuropathy. Acta Neuropathol
2008;115:231
–9. doi:10.1007/s00401-007-0326-2.
139. Tesch GH. Role of macrophages in complications of type 2
diabetes. Clin Exp Pharmacol Physiol 2007;34:1016
–9.
doi:
10.1111/j.1440-1681.2007.04729.x.
140. Conti G, Scarpini E, Baron P, Livraghi S, Tiriticco M, Bianchi R,
et al. Macrophage infiltration and death in the nerve during the
early phases of experimental diabetic neuropathy: a process
concomitant with endoneurial induction of IL-1beta and
p75NTR. J Neurol Sci 2002;195:35
–40. doi:10.1016/S0022-
510X(01)00684-0
.
141. Cameron NE, Cotter MA. Pro-inflammatory mechanisms in
diabetic neuropathy: focus on the nuclear factor kappa B
pathway. Curr Drug Targets 2008;9(1):60
–7, Jan.
142. Price SA, Zeef LA, Wardleworth L, Hayes A, Tomlinson DR.
Identification of changes in gene expression in dorsal root
ganglia in diabetic neuropathy: correlation with functional
deficits. J Neuropathol Exp Neurol 2006;65:722
–32.
doi:
10.1097/01.jnen.0000228199.89420.90.
143. Sullivan KA, Lentz SI, Roberts JL Jr, Feldman EL. Criteria for
creating and assessing mouse models of diabetic neuropathy. Curr
Drug Targets 2008;9:3
–13. doi:10.2174/138945008783431763.
144. Cohen CD, Klingenhoff A, Boucherot A, Nitsche A, Henger A,
Brunner B, et al. Comparative promoter analysis allows de novo
identification of specialized cell junction-associated proteins.
Proc Natl Acad Sci USA 2006;103:5682
–7. doi:10.1073/pnas.
0511257103
.
145. Schmid H, Cohen CD, Henger A, Irrgang S, Schlondorff D,
Kretzler M. Validation of endogenous controls for gene
expression analysis in microdissected human renal biopsies.
Kidney Int 2003;64:356
–60. doi:10.1046/j.1523-1755.2003.
00074.x
.
146. Schmid H, Henger A, Cohen CD, Frach K, Grone HJ,
Schlondorff D, et al. Gene expression profiles of podocyteassociated molecules as diagnostic markers in acquired proteinuric
diseases. J Am Soc Nephrol 2003;14:2958
–66. doi:10.1097/01.
ASN.0000090745.85482.06
.
147. Schmid H, Henger A, Kretzler M. Molecular approaches to
chronic kidney disease. Curr Opin Nephrol Hypertens
2006;15:123
–9. doi:10.1097/01.mnh.0000214770.11609.fb.