官网:http://grass.cgs.hku.hk/limx/kggseq/
kggseq软件是一款功能强大的测序后下游分析软件,现简要列举其功能如下,以备查询。
1. 质控(Quality Control)
-
对基因型的质控(Genotype QC)
参数如下:

-
对位点的质控(Variant QC)
参数如下:
 image.png
image.png
 image.png
image.png - 对样本的质控(Sample QC)
可以使用kggseq软件把vcf转换为plink格式进行质控,包括 (pairwise relatedness, mendel error rate and sex check)
java -jar kggseq.jar --vcf-file path-to-file1 --ped-file path/to/file2 --db-merge --o-plink-bed
plink --bfile kggseq.merged --genome
plink --bfile kggseq --mendel
plink --bfile kggseq --check-sex
- 也可以画MAF分布图
java -jar kggseq.jar --vcf-file path-to-file1 --out path/to/prefixname --mafplot
java -jar kggseq.jar --vcf-file path-to-file1 --ped-file path/to/file2 --db-filter hg18_dbsnp138,hg18_dbsnp141 --allele-freq 0,0.01 --o-vcf path/to/file3
2. 过滤(Filtering)
-
使用--genotype-filter参数,后面加上数字,数字对应意义如下:
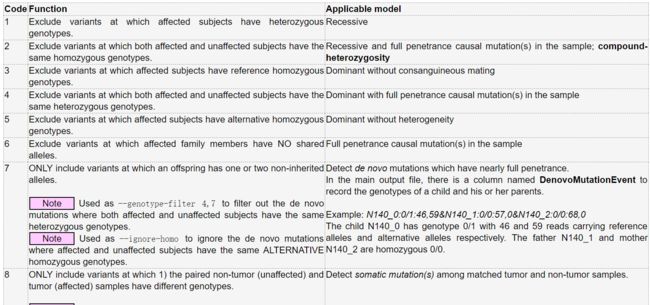 image.png
image.png
- 使用--double-hit-gene-trio-filter 或 --double-hit-gene-phased-filter参数提取复合杂合或者隐形突变位点
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --db-gene refgene --gene-feature-in 0,1,2,3,4,5,6 --db-filter 1kg201204,dbsnp137,ESP6500AA,ESP6500EA --rare-allele-freq 0.033 --db-score dbnsfp --mendel-causing-predict best --double-hit-gene-trio-filter (OR --double-hit-gene-phased-filter)
- 使用基因特征过滤(Gene feature filtering)
参数:--db-gene & --gene-feature-in (两个参数分别选择数据库和基因特征)
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --dgv-cnv-annot --splicing 6 --db-gene refgene,gencode,knowngene --gene-feature-in 0,1,2,3,4,5,6
基因特征代号如下:

image.png
- 使用公共数据库的基因型频率进行过滤(--db-filter)
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --db-filter 1kg201204,dbsnp141,ESP6500AA,ESP6500EA --rare-allele-freq 0.005 --db-filter-hard dbsnp138nf
也可以对本地数据进行频率过滤(--local-filter-hard)
- 过滤掉重复区域(--superdup-filter)或者注释这些区域(--superdup-annot)
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --superdup-filter
- 过滤掉indel,某些基因或某些区域
- --ignore-indel
- --ignore-snv
- --genes-out
- --regions-out
- 过滤掉假基因等其他因素造成的变异位点过多的基因(--gene-var-filter)
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 [options to filter out common and neutral variants] --gene-var-filter 4
- 过滤掉实验组/对照组独有的位点(--filter-model case-unique or --filter-model control-unique)
java -jar ./kggseq.jar --buildver hg19 --vcf-file path/to/file1 --ped-file path/to/file2 --filter-model case-unique
3. 注释(Annotation)
- 能注释的信息包括:
- SNP rs号(--rsid)
- 选择性剪切位点(--scsnv-annot)
- 该位点是否位于CNV区域(--dgv-cnv-annot)
- 该位点是否与感兴趣的基因在同一蛋白通路上(--candi-list gene1,gene2,...,geneN --ppi-annot ppiDatabase )
- 该位点是否与感兴趣的基因在同一基因通路上(--candi-list gene1,gene2,...,geneN --geneset-annot GenesetDatabase )
- 该位点最近的基因对应的小鼠表型(--mouse-pheno)
- 该位点最近的基因对应的斑马鱼表型(--zebrafish-pheno)
- 该位点最近的基因对应的疾病表型(--ddd-annot)
- 该位点最近的基因在pubmed数据库中的相关文献(--phenotype-term searchTerm and --pubmed-mining)
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --phenotype-term search+Term1,search+Term2 --pubmed-mining
PubMedIDIdeogram : PubMed ID of articles in which the term and the cytogeneic position of the variant are co-mentioned
PubMedIDGene : PubMed ID of articles in which the term and the gene containing the variant are co-mentioned
- 该位点对应基因在OMIM数据库中的疾病术语( --omim-annot)
- 该位点在COSMIC数据库中的注释信息(--cosmic-annot)
4. 位点功能的预测(Prediction at variant)
主要包括数据库:i.e., SIFT, PolyPhen2, GWAVA and CADD
- 预测孟德尔疾病致病位点(--db-score dbnsfp --mendel-causing-predict)
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --db-score dbnsfp --mendel-causing-predict best --filter-nondisease-variant
该方法通过对位点进行逻辑回归建模,评估前10个AUC曲线来确定候选位点。
也可以指定数据库进行打分预测(--mendel-causing-predict 4,6,7)
- 预测癌症中的高频体细胞突变(--db-score dbnsfp --cancer-mut-predict)
- 预测复杂疾病的致病位点
- 对非编码位点使用10个数据库进行打分预测(--db-score dbncfp_known [or dbncfp_all] --regulatory-causing-predict )
10个数据库包括:SuRFR、GWAS3D、FunSeq2、FATHMM-MKL、FunSeq、DANN、CADD_CScore、 GWAVA_Unmatched、GWAVA_TSS、GWAVA_Region。 - 通过添加细胞类型特异性调控甲基化分子标记预测致病基因(--db-score dbncfp_known [or dbncfp_all] --regulatory-causing-predict all --cell)
- 数据库资源包括(1)16 ENCODE cell types,默认采用GM12878细胞系。
(2)127 RoadMap human reference epigenomes ,默认采用E116 (GM12878 Lymphoblastoid Cell Line)
java -jar kggseq.jar --vcf-file path/to/file1 --db-score dbncfp_known --regulatory-causing-predict all --cell GM12878
java -jar kggseq.jar --vcf-file path/to/file1 --db-score dbncfp_known --regulatory-causing-predict all --cell E116
- 基因特征特异性模型(--db-score dbncfp_known [or dbncfp_all] --ldgf-func-predict)
用10个数据库的打分预测非编码区位点的致病情况
5. 针对基因的预测(Prediction at gene)
- 基因的致病性预测(--patho-gene-predict)
通过对Clinical Genomic Database数据库的打分,得到基因的致病打分。
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --out path/to/prefixname --excel --db-gene refgene --patho-gene-predict
- 表型挖掘(--phenotype-term searchTerm and --phenolyzer-prediction)
通过使用phenolyzer工具对表型相关基因进行排序,寻找到候选基因。
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --out path/to/prefixname --excel --db-gene refgene --phenotype-term searchTerm1,searchTerm2 --phenolyzer-prediction
- 组织特异性表达(--tissue-spec-annot)
使用GTEx数据库注释基因的表达情况。
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --out path/to/prefixname --excel --db-gene refgene --tissue-spec-annot
6. 统计检验
- 癌症驱动基因的负荷检验-ITER或者WITER或者WITER-ref方法(--iter-gene-coding)
java -jar witer.jar --maf-file path/to/file1.maf --out path/to/prefixname [other tags] --iter-gene-coding
java -jar witer.jar --maf-file path/to/file1.maf --out path/to/prefixname [other tags] --witer-gene-coding
java -jar witer.jar --maf-file path/to/file1.maf --out path/to/prefixname [other tags] --witer-gene-coding --ref-mut-file path/to/referenceDataset --cancer-label XXX
- 复杂疾病易感基因罕见突变位点负荷检验RUNER方法(--runer-gene-coding)
java -jar witer.jar --nt 6 [or more threads] --vcf-file path/to/file1 --ped-file path/to/file2 \
--out path/to/prefixname [or other tags] --excel --qqplot --gty-qual 20.0 --gty-sec-pl 20 --gty-dp 8 --gty-af-ref 0.05 \
--gty-af-het 0.25 --gty-af-alt 0.75 --ignore-indel --vcf-filter-in PASS --min-obs-rate 0.9 --hwe-all 0.001 \
--max-allele 3 --db-gene refgene,gencode --db-filter gadexome.[an ancestry tag],gadgenome.[an ancestry tag],1kgeas201305 \
--rare-allele-freq 0.01 --gene-freq-score [an ancestry tag] \
--db-score dbnsfp --disease-causing-predict best --runer-gene-coding
- 也可以选择编码区等位点进行分析( --response-gene-feature 2,3,4,5,6)
- 负荷检验后可以挖掘最相关的文献(--phenotype-term [disease name] --pubmed-mining-top-gene 10)
- 不将位点功能打分加入负荷检验模型(--uwruner-gene-coding)
- 使用本地control样本的频率进行检验(--gene-freq-score CONTROL)
- 加入其它协变量进行检验( --gene-cov-file),如:
Gene score1 score1 ...
SAMD11 2.114 0.076914032 ...
NOC2L 2.345 0.046528595 ...
KLHL17 1.989 0.026329064 ...
PLEKHN1 2.2 0.047853652 ...
... ... ... ...
7. 对位点的关联分析
- 卡方检验(--var-assoc)
java -jar ./kggseq.jar --buildver hg19 --var-assoc --p-value-cutoff 0.01 --multiple-testing benfdr --qqplot
- 使用 Rvtests软件的score, wald, exact, dominantExact, famLRT, famScore, famGrammarGamma 和 firth models等模型进行检验( --rvtest-var)
java -jar ./kggseq.jar --rvtest-var score,wald,exact,... --rvtest-vcf --rvtest-remove-set \
--vcf-file path/to/vcf/file --ped-file path/to/pedigree/file --phe phenotypeName \
--cov covariateName1,covariateName2,...,covariateNameN \
--out path/to/prefixname --excel --db-gene refgene
8. 对基因的关联分析
- 使用SKAT软件以基因为单位进行检验(--skat-gene)
包括SKAT,SKATO 和 Burden test算法。
java -jar ./kggseq.jar --skat-gene --vcf-file path/to/vcf/file --ped-file path/to/pedigree/file \
--phe phenotypeName --cov covariateName1,covariateName2,...,covariateNameN \
--out path/to/prefixname --excel \
--db-gene refgene [--p-value-cutoff 0.01 --multiple-testing benfdr --qqplot]
- 使用Rvtests(--rvtest-gene)软件进行检验
java -jar ./kggseq.jar --rvtest-gene cmc,cmat,price,skat[nPerm=1000:alpha=0.001:beta1=1:beta2=20],... --rvtest-vcf \
--rvtest-remove-set --vcf-file path/to/vcf/file --ped-file path/to/pedigree/file --phe phenotypeName \
--cov covariateName1,covariateName2,...,covariateNameN \
--out path/to/prefixname --excel --db-gene refgene
- 使用SKAT软件进行基因通路分析(--skat-geneset)
java -jar ./kggseq.jar --skat-geneset --geneset-file path/to/geneset/file --vcf-file path/to/vcf/file \
--ped-file path/to/pedigree/file --phe phenotypeName \
--cov covariateName1,covariateName2,...,covariateNameN \
--out path/to/prefixname --excel \
--db-gene refgene [--p-value-cutoff 0.01 --multiple-testing benfdr --qqplot]
基因通路需要存放在一个txt文件中,格式如下:
注:如果报错,可能是因为文件后面有空行,请用 --lib-update命令更新KGGSeq到最新版,或直接删除空行即可。
[Column 1: GeneSet ID]
[Column 2: GeneSet URL]
[Column 3..N: Gene Symbols in the geneset; separated by spaces].
No title row is required!!!
e.g.,
----------------------------------------------
KEGG_GLYCOLYSIS_GLUCONEOGENESIS http://www.broadinstitute.org/KEGG_GLYCOLYSIS_GLUCONEOGENESIS.html LDHC LDHB LDHA
KEGG_CITRATE_CYCLE_TCA_CYCLE http://www.broadinstitute.org/KEGG_CITRATE_CYCLE_TCA_CYCLE.html GJB1 OGDHL OGDH PDHB IDH3G LDHB
...
...
- 使用RVTEST软件进行基因通路分析(--rvtest-geneset)
最新版的 kggseq增加了参数--rvtest-min-var 2 可以去除位点小于2的基因。
java -jar ./kggseq.jar --rvtest-geneset cmc,skato[nPerm=1000:alpha=0.001:beta1=1:beta2=20],... --rvtest-vcf \
--rvtest-remove-set --geneset-file path/to/geneset/file --vcf-file path/to/vcf/file \
--ped-file path/to/pedigree/file --phe phenotypeName --cov covariateName1,covariateName2,...,covariateNameN \
--out path/to/prefixname --excel \
--db-gene refgene [--p-value-cutoff 0.01 --multiple-testing benfdr --qqplot]
- 基因富集分析(--geneset-enrichment-test)
查看给定的基因是否在某些给定的基因通路上排在前列
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --out path/to/prefixname [other tags] --geneset-enrichment-test
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --out path/to/prefixname --db-filter 1kg201204,dbsnp137,ESP6500AA,ESP6500EA --rare-allele-freq 0.01 --db-gene refgene --gene-mutation-rate-test --geneset-enrichment-test --geneset-db
可以自己设定通路基因(--geneset-file path/to/geneset/file)
9. 简单的画图功能
- MAF柱形图(reference dataset --mafplot-ref or in sample --mafplot-sample)
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --out path/to/prefixname --db-filter 1kg201204,dbsnp141,ESP6500AA,ESP6500EA --allele-freq 0,1 --mafplot-ref
java -jar kggseq.jar --vcf-file path/to/file1 --ped-file path/to/file2 --out path/to/prefixname --mafplot-sample
- QQ图
java -jar kggseq.jar --vcf-file path/to/file --out path/to/prefixname --qqplot --skat-gene [or] --var-assoc
10. 其他一些实用功能
- 计算LD和基因型相关性(--calc-ld)
java -Xmx15g -jar kggseq.jar --vcf-file path/to/file --ped-file path/to/file --out path/to/prefixname \
--calc-ld --db-filter 1kg201204,dbsnp141,ESP6500AA,ESP6500EA \
--allele-freq 0.05,0.95 --regions-in chr4:21212-233454
- 删除高LD的位点(--ld-prune X)
java -Xmx15g -jar kggseq.jar --vcf-file path/to/file \
--ped-file path/to/file --out path/to/prefixname \
--ld-prune 0.9 --filter-sample-maf-le 0.05 --o-svcf