刘小泽写于18.9.13
最近两天都在试着写GEO函数,想着做出来一起发送
但是好像没有那么简单,一步步正在调试
5 对结果进行注释~富集分析(基于超几何分布检验)
5.1 得到上调、下调基因
rm(list=ls())
load("GSE62832.DEG.Rdata")
DEG=DEG_mtx
if(T){
logFC_cutoff <- with(DEG,mean(abs(logFC)) + 2*sd(abs(logFC)) )
DEG$result = as.factor(ifelse(DEG$P.Value < 0.05 & abs(DEG$logFC) > logFC_cutoff,
ifelse(DEG$logFC > logFC_cutoff ,'UP','DOWN'),'NOT'))
}
gene <- rownames(DEG[DEG$result != 'NOT', ])
要再具体细分的话也可以
up_gene <- rownames(DEG[DEG$result =='UP',])
down_gene <- rownames(DEG[DEG$result =='DOWN',])
5.2 ID转换
使用clusterProfiler包接受的gene是Entrez ID,因此要先转换SYMBOL=》Entrez
几种主流的ID:
Ensemble id:由欧洲生物信息数据库提供,一般以ENSG开头,后边跟11位数字。如TP53基因:ENSG00000141510
Entrez id:由美国NCBI提供,通常为纯数字。如TP53基因:7157
Symbol id:为我们常在文献中报道的基因名称。如TP53基因的symbol id为TP53
Refseq id:NCBI提供的参考序列数据库:可以是NG、NM、NP开头,代表基因,转录本和蛋白质。如TP53基因的某个转录本信息可为NM_000546
library(clusterProfiler)
library(org.Hs.eg.db)
if(T){
gene_tr <- bitr(gene, fromType = "SYMBOL",
toType = c("ENSEMBL", "ENTREZID"),
OrgDb = org.Hs.eg.db)
up_gene_tr <- bitr(up_gene, fromType = "SYMBOL",
toType = c("ENSEMBL", "ENTREZID"),
OrgDb = org.Hs.eg.db)
down_gene_tr <- bitr(down_gene, fromType = "SYMBOL",
toType = c("ENSEMBL", "ENTREZID"),
OrgDb = org.Hs.eg.db)
gene <- gene_tr$ENTREZID
up_gene <- up_gene_tr$ENTREZID
down_gene <- down_gene_tr$ENTREZID
}
head(gene_tr)
#结果发现:总体有3.07%没有对应,上调有2.13%没有对应,下调3.76%没有对应
转换完可以去检验一下,比如检查Entrez ID: 23336, https://www.ncbi.nlm.nih.gov/gene/23336。确实得到的是SYNM这个基因
5.3 进行KEGG注释
最常见的就是GO/KEGG数据库注释,根据clusterProfiler,每个注释就对应一个函数
富集分析使用超几何分布检验:
比如背景基因这里有18837个,选出来的差异基因(上调235个+下调319个)554个,约占总体3%;
目前KEGG 包含了530个pathway,其中一个04110是Cell Cycle通路,其中包含124个基因【怎么统计?打开链接-》https://www.kegg.jp/dbget-bin/www_bget?hsa04110,找到gene栏-》使用linux cat命令变成一个列表 =〉wc】
如果是随机抽样的话,cell cycle在我们这554个基因中,应该能抽到124*3%=3个左右。也就是说,在得到的554个差异基因中,应该只有3个是cell cycle的基因;但结果找到了30个cell cycle的基因,那么就说明差异基因的产生都有cell cycle这个因素。如果是药物处理组的结果发现这样,就说明确实可能是药物起了作用,当然后续还需要结合其他分析
并非所有的通路都需要检测:有的通路比较小,只有十几个基因,我们这500多个差异基因中能抽到的理论上最多一个,这样一般就不考虑;还有的通路很大,有上千个基因,我们的背景基因才不到2w,那我们这586个差异基因中在这个pathway中的理论上就得有一百多个,这样理论和实际的变化差异就不明显,说明不了什么问题
enrichKEGG进行候选基因通路分析
if(T){
kk <- enrichKEGG(gene = gene,
organism = 'hsa',
pvalueCutoff = 0.05)
kk_gene <- (kk)[,1:6]
}
dotplot(kk,showCategory = 14,color="pvalue",font.size=14)
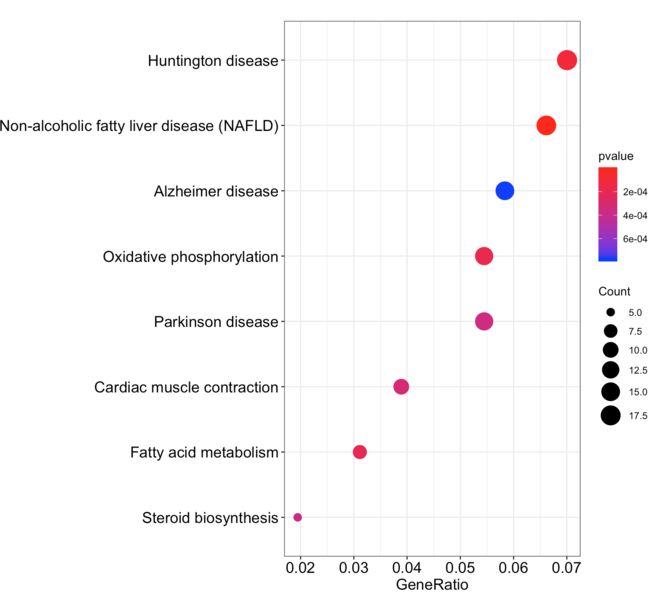
按照包的说明做一个geneList(标准:Entrez ID对应logFC,然后logFC降序排列)
if(T){
geneList <- DEG_mtx$logFC #先获得logFC【没有重复的18837个数值】
names(geneList) <- rownames(DEG_mtx) #获得基因名,但这里是SYMBOL格式【也是没有重复的18837个名称】
##问题就出在(下一步)SYMBOL转Entrez名字上,会多出来许多的重复:如果只是SYMBOL转Entrez,那么会得到18841个名称,原因就是一个symbol对应多个entrez ID,用count(geneList_tr,geneList_tr$SYMBOL) %>% arrange(desc(n))这样来查看到底哪些基因,结果查到有4个基因对应了2个entrez ID,其中一个是“HBD”,然后使用filter(geneList_tr, geneList_tr$SYMBOL == "HBD")来看HBD对应了哪个ID,果然发现HBD对应了两个ID;如果将SYMBOL转ENSEMBL+Entrez,那么会得到20198个名称,因为同一个SYNBOL,同一个Entrez,会有不同的Ensembl ID
#另外还有别人的友情提示:ENTREZID(具有唯一性):基因编号来自ANNOVAR的注释结果,建议别用SYMBOL,因为这种名称特异性较差,在转成ENTREZID时可能出现不唯一的现象。symbol与entrezID并不是绝对的一一对应的
geneList_tr <- bitr(names(geneList),
fromType = "SYMBOL",
toType = c("ENSEMBL","ENTREZID"),
OrgDb = org.Hs.eg.db)
#下一步目的:将Entrez ID与logFC联系起来
#因为转换后的名称已经不是和原来的geneList行名一一对应了,所以不能直接将原来SYMBOL行名变为Entrez ID。但是可以用SYMBOL名作为桥梁,一边连接logFC(做一个数据框),另一边连接Entrez ID(已经有的geneList_tr数据框),再将两个数据库按照SYMBOL联系起来
new_list <- data.frame(SYMBOL=names(geneList), logFC = as.numeric(geneList))
new_list <- merge(new_list, geneList_tr, by = "SYMBOL")
geneList <- new_list$logFC #重新生成geneList之logFC
names(geneList) <- geneList_tr$ENTREZID #重新生成geneList之Entrez ID
geneList <- sort(geneList,decreasing = T) #降序排列
}
gseKEGG GSEA分析
if(T){
kk2 <- gseKEGG(geneList = geneList,
organism = 'hsa',
nPerm = 1000,
minGSSize = 120,
pvalueCutoff = 0.2,
verbose = FALSE)
}
head(kk2)[,1:6]
gseaplot(kk2, geneSetID = "hsa04310")
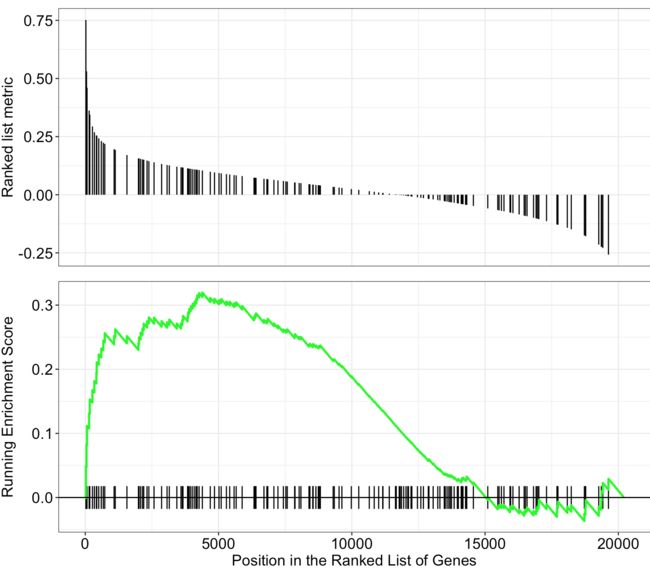
查看特定通路图
library(pathview)
hsa01212 <- pathview(gene.data = geneList,
pathway.id = "hsa01212", #上述结果中的hsa01212通路
species = "hsa",
limit = list(gene=max(abs(geneList)), cpd=1))
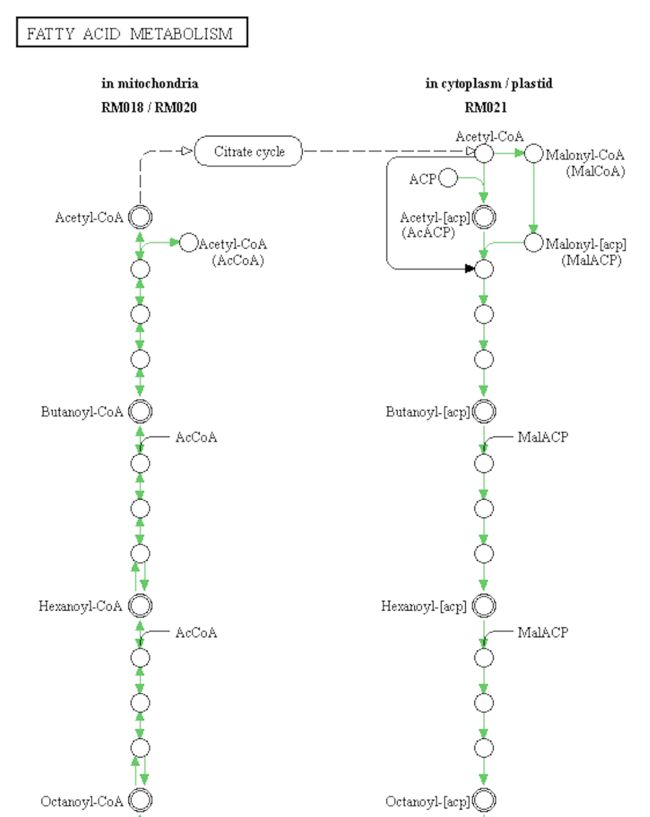
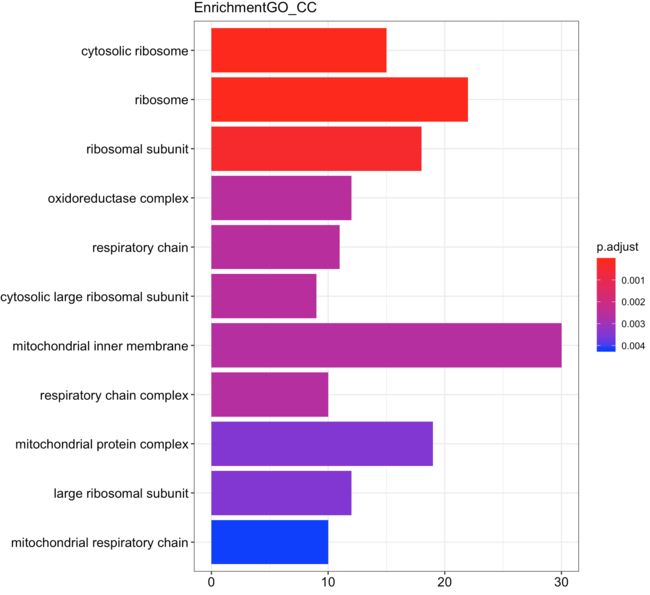
5.4 进行GO注释
if(T){
#这里以细胞组分CC为例
ego_CC <- enrichGO(gene = gene,
universe = names(geneList),
OrgDb = org.Hs.eg.db,
ont = "CC",
pAdjustMethod = "BH",
pvalueCutoff = 0.01,
qvalueCutoff = 0.05,
readable = TRUE)
}
ego_gene <- head(ego_CC)[,1:6]
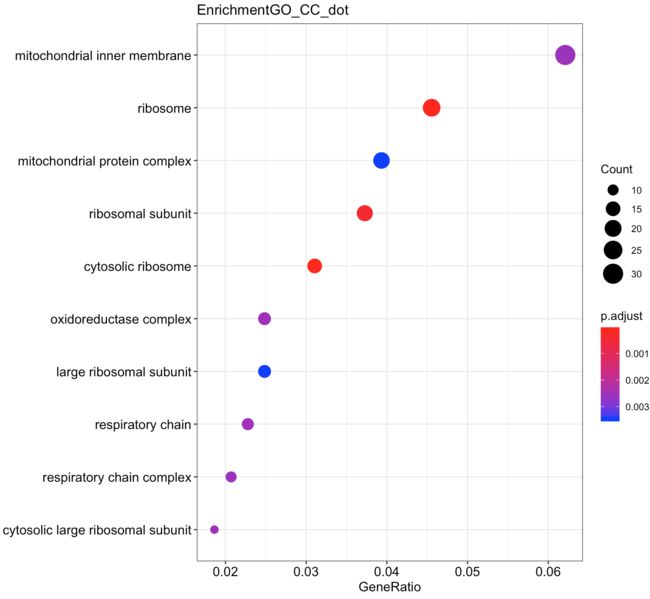
##可视化--点图
dotplot(ego_CC,title="EnrichmentGO_CC_dot")#点图,按富集的数从大到小的
##可视化--条形图
barplot(ego_CC, showCategory=20,title="EnrichmentGO_CC")#条状图,按p从小到大排,绘制前20个Term
#通路图
library(Rgraphviz)
library(topGO)
plotGOgraph(ego_CC)
欢迎关注我们的公众号~_~
我们是两个农转生信的小硕,打造生信星球,想让它成为一个不拽术语、通俗易懂的生信知识平台。需要帮助或提出意见请后台留言或发送邮件到[email protected]
