对所有生物过程来说,基因表达的精确调控对基因表达是非常重要的。序列特异性的转录因子结合在DNA 识别区域(cis调控元件),并且因此控制目标基因的转录起始速率,通过与其他比如co-factors的相互作用,染色体修饰和转录工厂等。人基因组编码大概1800个序列特异性转录因子,其中每一个都调控几百个目标基因。因为,TF 在基因表达中扮演重要角色,他们经常被认为是细胞过程的真正调控者。因此,这些TF的靶基因(target genes)的mapping和characterization对TF调控的生理过程可以提供非常重要的insight.例如,在癌症中,大概40%de driver 突变影响TF。并且许多关键的致癌和抑癌基因都是转录因子,比如p53,MYC,E2F,NF-KB等。在一个繁乱的基因网络中鉴定出转录因子,并且发现他们的目标基因,无论对于洞察肿瘤发生的过程还是对于治疗标的的发现都是非常重要的步骤。例如,虽然肿瘤抑制基因p53的很多靶基因都已经知道,但是p53下游的基因调控网络仍然不清楚。再比如,p53是否直接抑制靶基因仍然不清楚,是否p53和其他特殊的co-factors一起调控靶基因不清楚。是否不同的靶基因被调控依赖于不同的癌症类型或依赖于p53被激活的context情境?p53还是研究的比较多的,上面说的这种情况对其他研究的更少的转录因子情况更糟糕,有的转录因子到现在为止经常还没有靶基因被发现,有也很少。
已经知道的转录因子的靶基因可以通过实验确定,准确率还算比较高的,比如染色体免疫共沉淀实验followed by CHIP-Seq。但是这种方法也有缺陷,因为它通常只适用于细胞而非生物学样本(比如肿瘤)。并且,一次只聚焦一个单一的TF并且是先验的(a priori)。当转录因子不事先知道或者,只有基因表达谱可以执行,调控关系可以被揭示出来,通过基因表达普数据反推出来。解决这个问题的一个方式是,exploiting利用这样一个事实:被同一个TF调控的基因通常含有这个TF的结合位点。然后,对于人和这么庞大的基因组来说,若要在庞大的非编码区域鉴定这种短而易变的TF结合位点TF binding sites(TFBS),是一个计算挑战。虽然近年来很多motif 发现的发现出现,但单独这种方法对map一个基因调控网络来说是不够的,他们也不能被用于噪音基因集,这些基因集包含许多TF靶基因的集合。无论对于基于de novo的motif检测方法还是对于基于位置矩阵的伏击方法(PWM)都有这种缺点。另外一个限制因素,是许多方法被限制在使用已经注视的人PWMs(比如TRANSFAC,JASPAR,UNIPROBE).这就限制了基于motif富集方法鉴定候选网络TF的数目。所以,虽然顺式调控序列分析在解决TF-target相互作用上有潜能,但仍然有很多限制应用。
插件作者收集了多于九千个PWMs从不同的来源和物种,并且使用motif2TF程序,把它们和候选结合TFs联系在一起。并且作者开发了iregulon。允许,预测的cis调控结合位点直接整合进生物网络。最后,我们扩展和生成了这个框架,关于合并的motif,基于共表达基因集。
(暂且中断原来文章,因为牵扯知识相对深奥,所以先看一下这个插件的用法,然后继续翻译)http://iregulon.aertslab.org/tutorial.html
(暂且中断原来文章,因为牵扯知识相对深奥,所以先看一下这个插件的用法,然后继续翻译)http://iregulon.aertslab.org/tutorial.html
手册
iRegulon可以解决以下问题。
1。一个基因共表达网络的regulon是什么
2.在发表的signatures检测到的共同的靶基因是什么
3。共同调节microRNA靶基因的TF是什么
4.功能上成一聚类的基因比如同一个pathway的基因,直接的调控因子有什么
5.来自ChiP的靶基因的主要调控因子和辅助因子有什么
在一个共表达基因中预测regulons
这个例子里,iRegulon用于预测低氧处理的MCF7细胞上调的基因调控因子。原始的哦显示,HIF1A是低氧下的关键因子。作者使用了171个相关特征的基因列表名为“ELVIDGE_HYPOXIA_UP” in MSigDB (C2 CGP)
- 1.下载低压上调基因(我已经下载,在桌面)
- 2.点击file,import,network from table
- 3.selet 选择的file
- 4.选择column1作source interaction
-
5.点击import,a network of 171 nodes会被创建
 image.png
image.png
2.query iRegulon 预测regulators和靶基因
Query格式允许用户给这个分析一个名字。可以定义基因名称,可以定义database来询问,motif collection,搜索space的类型(基于基因还是基于区域),调控的搜索区域(TSS上游500bp,TSS周围10kb或20kb)还有保守型(7或10个物种)。Motif预测参数是伏击分数阈值,ROC阈值(AUC算法),还有靶基因选择的rank 阈值。
- 1)选择网络所有nodes
- 2)点击apps,iregulon,predict regulators and targets这个时候不一定刚才输入的所有170个基因都有效。可能会出现163/171 genes are valid(备注,这个在新版本中没有这一项了,另外因为数据更新,有些数据变了,比如motif collection,以后还会增加。还有,界面也变了,下面我显示的是最新界面20170211)
-
3)所有的其他值用默认值,点submit
 image.png
image.png
3.结果
首先记得,result panel可以点击最大
以前是有两个选项卡,现在有三个。(我暂且还不知道什么意思,以后慢慢添加)
先看motif-centric view
这是最原始的结果,是一个富集motif的列表,针对可以结合到这个motif上的候选转录因子都有一个排序。主面板显示的是根据NES的排序,得分越高,越靠前。另外还有AUC,预测的targets的数目(#targets) ,还有TFs的数目(这个是根据motif2TF算法来的)。注意,假如TFs的数目是0,这意味着这个motif不能和已经知道的TF联系,但仍然可以被预测是富集的。下面的表是和被选择的某个motif有关(蓝色背景高亮)。并且,左边是,相联系的TFs(有evidence 参数值,motif相似度和%identity)2.右边,这个motif相应的预测的靶基因(有他们的排序)
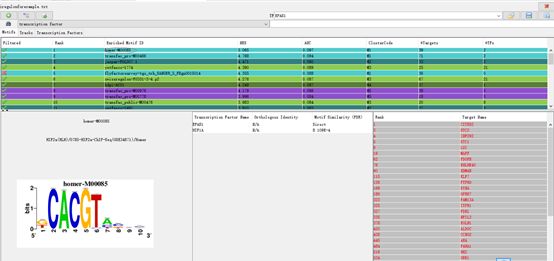
注意这个结果和作者原来结果不一样了,第一个成了homer-M00085估计是新发现的motif
再看Transcription factors选项卡 TF-centric view
上面这个表显示的是根据NES排序的富集TFs排名。合并后的targets数目( Targets)有显示,当然这是根据很多motif发现的(#motifs)。下面的表显示的是上边选定的某个TF的motifs结果,中间这个表显示的预测的TFs根据motif2TF可以和这些motif联系在一起的证据是水平(%identity,motif similarity和motif的数目)右边的表显示的是,targets的排序和预测的和他们相关的motifs的数目
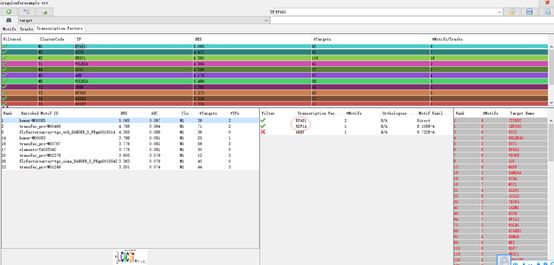
注意这个表已经和作者当时的不一样了。
得分最高的regulon和HIF1A富集分数4.743相符合,(来自13个motifs),有97个在hypoxia中上调的靶基因。
The top scored regulon corresponds to the HIF1A enriched with a score of 4.743(resulting from 13 motifs)with 97 direct targets up-regulated in hypoxia.
预测metatargetomes
给定的一个TF,预测其靶基因
这个例子里,我们预测TP53的靶基因组(TP53是一个抑癌基因,在癌症中经常发生突变)
1.query the metatargetome
1)点击apps ,iregulon,query cistargetDB
2)查询格式允许用于选择TF,物种和geneset数据库(genesigDB,ganesh cluster,MSigDB). 下面一些参数可以设置比如最大最小值等

3)一般选择好TF直接submit就可以了,其余默认值即可
2.结果展示
以网络形式展示结果
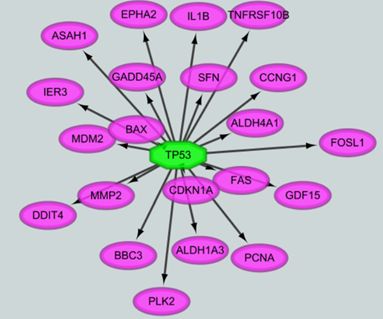
现在的展示结果和以前不一样了,regulator(TP53)是八边形,靶基因是椭圆形紫色。(iRegulon visual style in vizmapper默认).
预测TF-miRNA共调控
若应用与miRNA靶基因,ireglon揭示TF和miRNA regulon之间的交叉
在这个例子里,我们看miRNA-133a,这是在肌肉组织表达的,看起来可以抑制非肌细胞基因的表达
1. 下载miRNA targetome
1)去miRWalk网站http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/
2)validated targets ,microRNA targets
3)选择humans for species
4)输入has-mir-133a
5)search
6)结果页面的底部,点击download table 下载has-mir-133a已知的targetome
2. run iregulon
1)转换excel文件格式成tab-delimited format(.txt)
2)点击file,import,network from table
3)select file,选中
4)column1(microRNA)source interaction
5)column4(target gene name)for target interaction
6)tick show text file import options to define the start import row if necessary
7)import, network会被建立。
8)选择所有nodes
9)apps,iregulon,predict regulators and targets
10)所有默认即可,submit
3.结果显示
Detect功能相关基因的直接调控关系(如同一信号通路)
对于一个给定的功能相关基因(例如一个信号通路里的),iregulon可以鉴定调控因子和TF-target相互关系。
在这个例子中,我们聚焦notch信号通路,来鉴定已经知道的参与notch信号通路的调控因子(RBPJ,HEY1,HEY2,HES1)
1.得到参与notch信号通路的基因
为了得到这些基因,我们使用pathway commons web service client from cytoscape to import the “notch signaling pathway” from NCI/Nature pathway interaction database(ID:notch_pathway)(注意这里 在新版cytoscape变了)
1)file,import,network from public databases
2)use the wikipathways
3)enter notch, search
4)select notc1
5)双击“notch signaling pathway”
2. run iregulon
1)选择所有nodes
2)iregulon,predict regulators and targets
3)同上
3结果展示
1)transcription factor view,
2)在前面的tf中,HES1,RBPJreglulon是notch信号通路重要调节因子
虽然用的数据和作者用的不一样,但结果一致。

基于chip targets的co-factors发掘