接着上篇继续练习,前一篇文章里已经把PBMC按照4个时间点分了群,接下来就是差异分析了。本次使用monocle进行差异分析,主要分10步(使用monocle做拟时序分析(单细胞谱系发育)):
step1: 创建对象
step2: 质量控制
step3: 表达量的标准化和归一化
step4: 去除干扰因素(多个样本整合)
step5: 判断重要的基因
step6: 多种降维算法
step7: 可视化降维结果
step8: 多种聚类算法
step9: 聚类后找每个细胞亚群的标志基因
step10: 继续分类
(六)差异分析 (monocle V2)
这部分将按照教程(10X scRNA免疫治疗学习笔记-5-差异分析及可视化)来练习。我下载的是2017年版本monocle V2。其功能也不仅仅是差异分析那么简单。还包括pseudotime,clustering分析,而且还可以进行基于转录本的差异分析(单细胞转录组3大R包之monocle2)。
准备数据
文献中是对PBMC_RespD376这一个子集做了差异分析,所以我也用这个子集来练习。
上面已经取出了PBMC_RespD376这个子集:
> PBMC_RespD376=SubsetData(PBMC,cells=([email protected]$TimePoints =='PBMC_RespD376'))
> table([email protected])
0 1 2 3 4 5 6 7 8 9 10 11 12 13
806 747 527 717 339 459 90 285 71 202 220 68 135 18
接下来文章主要分析了CD8+ T细胞的差异表达,CD8+ T细胞分成了两群:红色(编号10)和浅蓝色(编号4),所以需要提取出这两个群的细胞:
> PBMC_RespD376_for_DEG = SubsetData(PBMC_RespD376, ident.use =c(4,10))
利用monocle V2构建S4对象
使用其提供的 newCellDataSet() 函数构建对象(继承自ExpressionSet对象),对象组成成分(一些单细胞转录组R包的对象):
(1)表达量矩阵exprs:数值矩阵。行名是基因, 列名是细胞编号。
(2)细胞的表型信息phenoData: 第一列是细胞编号,其他列是细胞的相关信息。
(3)基因注释featureData: 第一列是基因编号, 其他列是基因对应的信息。
那ExpressionSet对象到底是什么?下面这张图可以直观的说明:

并且这三个数据集要满足如下要求:
表达量矩阵必须:
(1)保证它的列数等于phenoData的行数
(2)保证它的行数等于featureData的行数)
而且:
(4)phenoData的行名需要和表达矩阵的列名匹配。
(5)featureData和表达矩阵的行名要匹配。
(6)featureData至少要有一列"gene_short_name", 就是基因的symbol。
简单的了解了对象,接下来看一下在上面取出的子集里,我们可以看看Seurat object里面都有什么:

在这图里面,@meta.data一般会存放表型相关的信息如cluster、sample的来源、group等; @assay中的@counts存放单细胞测序的raw data,data储存了归一化后的数据,而scale.data存储了标准化之后的数据(一般是z-score)。课程里使用的是归一化后的(实际上我认为需要使用最原始的,也就是counts里的数据,理由见下面的分析。但是由于为了跟课程一致,我这里用了和课程一样的归一化后的数据),需要注意的是,不要用scale.data,也就是不要用标准化之后的data来构建monocle对象,官网是这样说的:

然后开始做准备工作:
#准备表达矩阵(monocle需要的表达量矩阵exprs)
> count_matrix=PBMC_RespD376_for_DEG@assays[["RNA"]]@data
> dim(count_matrix)
[1] 17712 559
# 细胞分群信息
> [email protected]
> table(cluster)
cluster
0 1 2 3 4 5 6 7 8 9 10 11 12 13
0 0 0 0 339 0 0 0 0 0 220 0 0 0
#细胞名称(就是barcode)
> names(as.data.frame(count_matrix))
[1] "AAAGTAGTCTGCTGTC.3" "AACACGTAGATGCGAC.3" "AACCATGAGGCTCTTA.3" "AACCATGCAGGCTCAC.3" "AACCGCGCAGCGAACA.3"
[6] "AACTCTTGTTTACTCT.3" "AACTTTCAGAGTGAGA.3" "AACTTTCGTATAGGGC.3" "AAGACCTAGCTAGTCT.3" "AAGCCGCAGGGTTCCC.3"
[11] "AAGGAGCTCCTTGGTC.3" "AAGGCAGCAGTTAACC.3" "AATCGGTAGCCACTAT.3" "ACACCCTGTCTCTCTG.3" "ACAGCTAAGATCCTGT.3"
......
#基因信息
> gene_annotation <- as.data.frame(rownames(count_matrix))
以上就是所有monocle需要的东西,下面就是开始monocle分析了:
# 构建monocle需要的表达量矩阵exprs
> expr_matrix <- as.matrix(count_matrix)
# 构建monocle需要的细胞表型信息phenoData
> sample_ann <- data.frame(cells=names(as.data.frame(count_matrix)),
cellType=cluster)
> rownames(sample_ann)<- names(as.data.frame(count_matrix))
# 构建monocle需要的基因注释featureData
> gene_ann <- as.data.frame(rownames(count_matrix))
> rownames(gene_ann)<- rownames(count_matrix)
> colnames(gene_ann)<- "genes"
# 转换成AnnotatedDataFrame需要的对象
> pd <- new("AnnotatedDataFrame",
data=sample_ann)
> fd <- new("AnnotatedDataFrame",
data=gene_ann)
# 开始构建CDS对象
> sc_cds <- newCellDataSet(
expr_matrix,
phenoData = pd,
featureData =fd,
expressionFamily = negbinomial.size(),
lowerDetectionLimit=1)
> cds=sc_cds
> cds <- detectGenes(cds, min_expr = 0.1)
这里提一下expressionFamily这个参数,这个参数有几种可以选择的value:
我们用的是第一种negbinomial.size(),官网上说第二种比第一种要更好一些,但是缺点就是特!别!慢!,适用于那些非常小的数据。


# 结果在cds@featureData@data里
# 看一下里面都有什么:
> print(head(cds@featureData@data))
genes num_cells_expressed
RP11-34P13.7 RP11-34P13.7 0
FO538757.2 FO538757.2 14
AP006222.2 AP006222.2 6
RP4-669L17.10 RP4-669L17.10 0
RP11-206L10.9 RP11-206L10.9 7
LINC00115 LINC00115 0
CDS对象构建完成之后,需要过滤一下,
> expressed_genes <- row.names(subset(cds@featureData@data,
+ num_cells_expressed >= 5))
> length(expressed_genes)
[1] 7718
> cds <- cds[expressed_genes,]
monocle聚类
第一步:挑选细胞进行聚类(挑选的标准一般是表达量不要太低,最好是差异表达基因)
> cds <- estimateSizeFactors(cds)
> cds <- estimateDispersions(cds)
Removing 6 outliers
> disp_table <- dispersionTable(cds) # dispersionTable() 函数用来挑选有差异的基因
> unsup_clustering_genes <- subset(disp_table, mean_expression >= 0.1) # 挑表达量不太低的
> cds <- setOrderingFilter(cds, unsup_clustering_genes$gene_id) # 准备聚类基因名单
> plot_ordering_genes(cds)

第二步:选主成分
> plot_pc_variance_explained(cds, return_all = F)

第三步:聚类
先降维:
> cds <- reduceDimension(cds, max_components = 2, num_dim = 6,
reduction_method = 'tSNE', verbose = T)
Remove noise by PCA ...
Reduce dimension by tSNE ...
再聚类:
> cds <- clusterCells(cds, num_clusters = 4)
Distance cutoff calculated to 1.506418

monocle差异分析
先提取差异基因:
> start=Sys.time()
> diff_test_res <- differentialGeneTest(cds,
+ fullModelFormulaStr = "~cellType")
> end=Sys.time()
> end-start
Time difference of 2.261041 mins
> sig_genes <- subset(diff_test_res, qval < 0.1)
> nrow(sig_genes)
[1] 545
> head(sig_genes[,c("genes", "pval", "qval")] )
genes pval qval
ISG15 ISG15 5.108942e-05 2.075306e-03
TNFRSF4 TNFRSF4 4.142010e-04 1.206341e-02
RPL22 RPL22 3.093532e-03 5.657792e-02
PARK7 PARK7 1.259435e-09 1.869292e-07
ENO1 ENO1 6.332727e-04 1.696358e-02
EFHD2 EFHD2 5.721690e-04 1.571530e-02
(七)可视化(热图)
在文献中,作者挑选了如下基因进行可视化:
> htmapGenes=c(
'GAPDH','CD52','TRAC','IL32','ACTB','ACTG1','COTL1',
'GZMA','GZMB','GZMH','GNLY'
)
先来看看这些基因是不是也在我前面分析得到的差异基因里:
> htmapGenes %in% rownames(sig_genes)
[1] TRUE TRUE FALSE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
貌似第三个基因不在我的差异分析结果里,所以把第三个基因去除掉,
> htmapGenes=c(
'GAPDH','CD52','IL32','ACTB','ACTG1','COTL1',
'GZMA','GZMB','GZMH','GNLY'
)
> htmapGenes %in% rownames(sig_genes)
[1] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
下面开始画图:
library(pheatmap)
dat=count_matrix[htmapGenes,]
pheatmap(dat,show_colnames =F,
show_rownames = T,
cluster_cols = F,
cluster_rows = F)
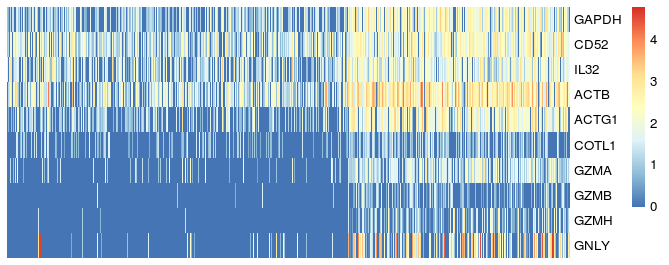
感觉不是很明显,把代码修饰一下,
> n=t(scale(t(dat)))
# 根据原文中的来限定热图的scale上限和下限,意思是大于2的都按照2来处理,小于-1的都按照-1来处理:
> n[n>2]=2
> n[n< -1]= -1
> n[1:4,1:4]
AAAGTAGTCTGCTGTC.3 AACACGTAGATGCGAC.3 AACCATGAGGCTCTTA.3 AACCATGCAGGCTCAC.3
GAPDH -0.3703769 -0.5844036 -1.0000000 -0.01595905
CD52 -0.7797171 -0.4864851 -1.0000000 0.33213334
IL32 1.4325850 -1.0000000 0.2037381 1.80063967
ACTB -1.0000000 -0.5737242 1.1705545 0.50459707
然后加上细胞的群的cluster名字4和10:
> ac=data.frame(group=cluster)
> rownames(ac)=colnames(n)
下面再画一个试试:
> pheatmap(n,annotation_col = ac,
show_colnames =F,
show_rownames = T,
cluster_cols = F,
cluster_rows = F)

###############################我是分割线#################################
针对GSE117988的tumor部分进行分析
上面的分析以及前一篇的分析都是基于这个病人的PBMC(外周血单核细胞)的,下面就来对tumor的部分进行分析。原始矩阵下载地址:https://www.ncbi.nlm.nih.gov//geo/query/acc.cgi?acc=GSE117988。代码按照教程10X scRNA免疫治疗学习笔记-6-marker基因的表达量可视化的步骤走。
(一)读取数据
> raw_dataTumor <- read.csv('./GSE117988_raw.expMatrix_Tumor.csv.gz', header = TRUE, row.names = 1)
> dim(raw_dataTumor)
[1] 21861 7431
这里肿瘤样品分为两种:治疗前(Pre),复发(AR)。
(二)Seurat常规步骤
(1)归一化
> dataTumor <- log2(1 + sweep(raw_dataTumor, 2, median(colSums(raw_dataTumor))/colSums(raw_dataTumor), '*'))
> head(colnames(dataTumor))
[1] "AAACCTGAGGATGTAT.1" "AAACCTGCAGCGATCC.1" "AAACCTGGTACGAAAT.1" "AAACGGGAGCTGGAAC.1" "AAACGGGAGGAGTTGC.1"
[6] "AAACGGGAGTTTAGGA.1"
(2)划分细胞类型
#这一步我用的是和之前一样的unlist,而教程用的是一个新函数ExtractField,在我的R studio里这个函数是报错的,所以我还是换用了之前的代码。
> cellTypes <- sapply(colnames(dataTumor), function(x) unlist(strsplit(x, "\\."))[2])
> cellTypes <-ifelse(cellTypes == '1', 'Tumor_Before', 'Tumor_AcquiredResistance')
> table(cellTypes)
cellTypes
Tumor_AcquiredResistance Tumor_Before
5188 2243
#结果和教程里的一样
(3)对表达矩阵进行质控
这一步和之前是一样滴~
# 基因在多少细胞表达
> fivenum(apply(dataTumor,1,function(x) sum(x>0) ))
VP2 GPRIN2 EML3 ZNF140 RPLP1
1 8 103 566 7431 #75%的基因在566个细胞中表达
# 一个细胞中有多少表达的基因
> fivenum(apply(dataTumor,2,function(x) sum(x>0) ))
GGAACTTAGGAATCGC.1 TACGGTACAAGCCGCT.2 CCTACCAAGCGTGAGT.1 TGAGCCGAGACTAGAT.2 GATCGTAGTCATATGC.2
192.0 1059.0 1380.0 1971.5 5888.0
##基本上细胞里有1000个基因的表达
(4)创建Seurat对象
> tumor <- CreateSeuratObject(dataTumor,
+ min.cells = 1, min.features = 0, project = '10x_Tumor')
> tumor
An object of class Seurat
21861 features across 7431 samples within 1 assay
Active assay: RNA (21861 features)
(5)添加metadata
> tumor <- AddMetaData(object = tumor, metadata = apply(raw_dataTumor, 2, sum), col.name = 'nUMI_raw')
> tumor <- AddMetaData(object = tumor, metadata = cellTypes, col.name = 'cellTypes')
(6)聚类
> tumor <- ScaleData(object = tumor, vars.to.regress = c('nUMI_raw'), model.use = 'linear', use.umi = FALSE)
Regressing out nUMI_raw
|================================================================================================================| 100%
Centering and scaling data matrix
|================================================================================================================| 100%
> tumor <- FindVariableFeatures(object = tumor, mean.function = ExpMean, dispersion.function = LogVMR, x.low.cutoff = 0.0125, x.high.cutoff = 3, y.cutoff = 0.5)
Calculating gene variances
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
Calculating feature variances of standardized and clipped values
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
> tumor <- RunPCA(object = tumor, pc.genes = [email protected])
PC_ 1
Positive: STMN1, HNRNPA2B1, CBX3, TUBB2B, PHF14, HMGN2, NDUFA4, SUB1, UCHL1, TUBA1A
MDK, PCDH9, ZFAS1, ILF2, CALM2, PRDX2, DEK, CCER2, SOX4, NEFM
CCT5, MARCKSL1, TUBB, KRT18, NHLH1, HMGB2, CCT6A, CKB, HIST1H4C, HSP90AB1
Negative: CD74, HLA-DRA, S100A11, HLA-DPB1, HLA-DRB1, TYROBP, HLA-DPA1, VIM, LGALS1, C1QB
SAT1, C1QA, SRGN, CTSB, NPC2, SOD2, FCER1G, CD68, APOE, HLA-DQB1
HLA-B, HLA-DQA1, APOC1, LAPTM5, CTSS, HLA-DRB5, AIF1, C1QC, PSAP, TYMP
PC_ 2
Positive: SELM, MT-CO3, CD44, MT-ATP6, S100A1, S100A16, MYO15A, MT-CYB, RBP7, LGALS7B
MT-ND3, NEAT1, CCER2, MT-ND2, CYBA, PSTPIP1, PCP4, ATP5J, ADAMTS7, PHLDA2
RPL22L1, HIST1H1C, PGF, IGKC, POLR2L, H1F0, CDKN1A, SH3BGRL3, S100A2, TFAP2B
Negative: HSP90AA1, ACTB, HNRNPA2B1, TUBA1A, HSPA8, ACTG1, HSP90AB1, TUBA1B, HSPD1, TUBB2B
CALM2, DNAJA1, BASP1, HMGB2, UBB, UBC, SUB1, VAPA, DNAJB6, HSPH1
KPNA2, HSPB1, CKS2, HSPA1A, CACYBP, PCDH9, UBE2S, CCT5, H3F3B, PTTG1
PC_ 3
Positive: IGFBP7, IFITM3, SPARC, CALD1, MGP, NNMT, SPARCL1, TM4SF1, EMP1, IFI27
CAV1, LAMA4, IGFBP4, IFITM2, COL4A2, PTRF, TIMP1, COL4A1, COL3A1, COL1A1
BGN, PRSS23, C1R, COL6A2, CCL2, COL1A2, GNG11, FSTL1, FN1, ZFP36L1
Negative: C1QB, C1QA, TYROBP, FCER1G, CD68, AIF1, APOC1, C1QC, HLA-DQA1, LYZ
HLA-DQB1, CTSB, C15orf48, LAPTM5, ITGB2, GPR183, CAPG, HLA-DMB, APOE, HLA-DPA1
CTSD, C5AR1, PCP4, SDS, LST1, LIPA, MS4A7, ACP5, HLA-DRA, MS4A6A
PC_ 4
Positive: HSPA1A, ZFAND2A, DNAJB1, HSPB1, HSPA1B, HSPH1, HSPA6, ATF3, SERPINH1, JUN
CHORDC1, SNHG15, DEDD2, DNAJA4, CLK1, MRPL18, DNAJB6, HSP90AB1, RSRC2, HSPD1
CACYBP, FKBP4, SNHG8, IER2, HSP90AA1, BAG3, DNAJA1, TCP1, ZFAS1, HERPUD1
Negative: HMGN2, UBE2C, H2AFZ, PTTG1, STMN1, CALM2, BIRC5, CKS1B, UBE2S, HMGB1
CENPF, HMGB2, TMSB10, TOP2A, CENPA, MKI67, CCNB2, TUBB4B, GSTP1, HN1
HIST1H4C, ASPM, CCNB1, PRDX2, NUF2, NDUFA4, NUSAP1, TMSB4X, PCP4, TUBA1B
PC_ 5
Positive: TOP2A, UBE2C, CENPF, CKS1B, BIRC5, ASPM, CCNB1, CCNB2, MKI67, NUF2
CENPA, NUSAP1, PIF1, SMC4, KPNA2, CKS2, HMGB2, PTTG1, TUBA1C, ARL6IP1
HIST1H4C, UBE2S, NUCKS1, IGKC, TUBB4B, CD44, JUN, ATF4, ISG20, CREB5
Negative: CHGA, VIP, NPY, CD63, MDK, KRT18, PCSK1N, ENO1, KRT8, ALDOA
NDUFS6, TPI1, NDUFA4, CCER2, SUB1, UCHL1, CCK, NDUFB2, SEC61G, PRDX2
S100A6, MARCKSL1, FTL, CKB, CST3, MYL6, PGK1, GSTP1, NDUFV2, PCDH9
> tumor <- RunTSNE(object = tumor, dims.use = 1:10, perplexity = 25)
> DimPlot(tumor, group.by = 'cellTypes', colors.use = c('#EF8A62', '#67A9CF'))
#最后把这个Seurat对象保存下来
> save(tumor,file = 'patient1.Tumor.V2.output.Rdata')

(三)基因可视化
> load('patient1.Tumor.V2.output.Rdata')
#取归一化后的表达矩阵,储存在data里
> count_matrix=tumor@assays[["RNA"]]@data
> count_matrix[1:4,1:4]
4 x 4 sparse Matrix of class "dgCMatrix"
AAACCTGAGGATGTAT.1 AAACCTGCAGCGATCC.1 AAACCTGGTACGAAAT.1 AAACGGGAGCTGGAAC.1
VP2 . . . .
largeTAntigen 0.9670525 . . .
smallTAntigen . . . .
RP11-34P13.7 . . . .
#取出细胞分群信息
> [email protected]$cellTypes
> table(cluster)
cluster
Tumor_AcquiredResistance Tumor_Before
5188 2243
下面是提取基因信息,在文献里,作者针对治疗前和复发组的HLA-A和HLA-B的变化。首先要先看一下这两个基因是不是在data里
> allGenes = row.names(tumor@assays[["RNA"]]@counts)
> allGenes[grep('HLA',allGenes)]
[1] "HHLA3" "HLA-F" "HLA-G" "HLA-A" "HLA-E" "HLA-C" "HLA-B" "HLA-DRA" "HLA-DRB5" "HLA-DRB1"
[11] "HLA-DQA1" "HLA-DQB1" "HLA-DQA2" "HLA-DQB2" "HLA-DOB" "HLA-DMB" "HLA-DMA" "HLA-DOA" "HLA-DPA1" "HLA-DPB1"
看到这两个基因确实在里面,然后就可以画图了,首先对HLA-A操作:
> FeaturePlot(object = tumor,
features='HLA-A',
cols = c("grey", "blue"),
reduction = "tsne")
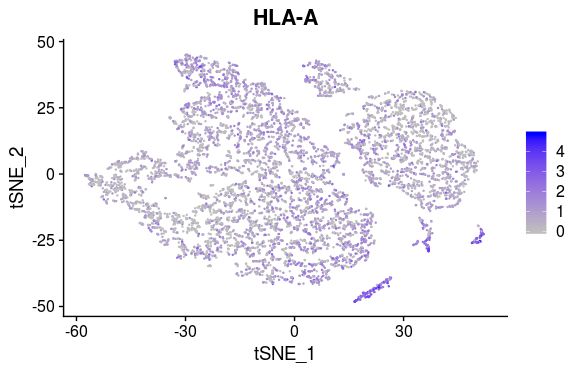
HLA-A的表达量:
> table(count_matrix['HLA-A',]>0, cluster)
cluster
Tumor_AcquiredResistance Tumor_Before
FALSE 2282 1057
TRUE 2906 1186
#在AR组里有2906个细胞表达HLA-A,在治疗前组里有1186个细胞表达这个基因
再对HLA-B画图:
> FeaturePlot(object = tumor,
features ='HLA-B',
cols = c("grey", "blue"),
reduction = "tsne")

#HLA-B的表达量
> table(count_matrix['HLA-B',]>0, cluster)
cluster
Tumor_AcquiredResistance Tumor_Before
FALSE 4794 1258
TRUE 394 985
#可以看出对于HLA-B这个基因在两个组里表达的细胞数量都很少,另外如果横向比较的话,治疗前表达比复发组要高。
总结:
重复了文献里的一些图,走了一遍10×单细胞测序分析过程。这个过程的收获就是接触到了单细胞分析的另一个包monocle2,也是很流行的一个包。因为走流程走的比较快,对里面的一些细节还需要深入的学习。monocle的官方网站http://cole-trapnell-lab.github.io/monocle-release/docs/里面有非常详细的步骤和很多注意事项,值得深入的学习一遍。