欢迎关注”生信修炼手册”!
MACS是一款最为流行的peak calling软件,最初是针对转录因子的chip数据来设计的,在最新版本中,也添加了对组蛋白修饰的适配。目前最新版本为v2.0,官网如下
https://github.com/taoliu/MACS
在2.0版本中提供了以下多个子命令
callpeak
bdgpeakcall
bdgbroadcall
bdgcmp
bdgopt
cmbreps
bdgdiff
filterdup
predictd
pileup
randsample
-
refinepeak
每个子命令和对应的功能描述如下

本文主要介绍macs2最经典的使用场景peak calling, 基本用法如下
macs2 callpeak \
-t ip.bam \
-c input.bam \
--outdir out_dir \
-n chip \
-g hs-t参数指定抗体处理的样本,-c指定input样本,值得一提的是,macs支持多种格式的输入文件,除了上述代码中使用的bam格式外,还支持SAM/BED格式。
--outdir指定输出结果的目录,-n参数指定输出文件名的前缀,-g参数指定基因组的有效大小,在NGS数据中,测序reads在基因组上的覆盖度并不是100%, 而且有些重复区域的比对信息是不可信的,剩下的能够利用的区域通常只占整个基因组区域的70%到90%,这个区域的大小就是有效大小,对于常见的物种,程序内置了有效大小,我们只需要指定物种的缩写即可

对于其他物种,则需要自己指定有效基因组的大小,单位为bp。
输出文件如下
chip_model.r
chip_peaks.narrowPeak
chip_peaks.xls
chip_summits.bedmodel.r是一个可执行的R脚本,通过以下代码可以产生一个PDF的输出文件
Rscript chip_model.r第一页表示peak邻近区间正负链测序分布,用于评估d这个参数值,示意如下
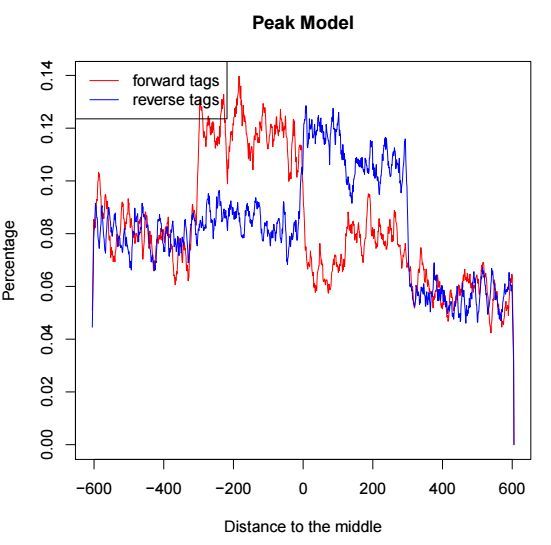
第二页是cross-correlation分析的结果,示意如下

后缀为xls的文件是peak的输出结果,内容示意如下

#开头的是注释信息,显示了软件调用的具体命令和参数设置,便于核查;其他的行记录了peak的区间信息,这里的起始位置采用的是从1开始计数的方式。
后缀为narrowpeak的文件是一个BED格式的文件,内容示意如下

前四列代表peak区间和名称,注意bed格式中起始位置从0开始计数,第五列的值为int(-10*log10qvalue),第六列全部为.,第七列为fold_enrichment,第八列为-log10(pvalue),第九列为-log10(qvalue),第十列为peak的中心,即summit距离peak起始位置的距离,对应abs_summit - start。
后缀为bed的文件为peak中心,即summit对应的bed文件,内容示意如下

最后一列为-log10(qvalue)。以上就是macs2 peak calling的基本用法,更多详细的参数和用法请参考官方文档。
·end·
—如果喜欢,快分享给你的朋友们吧—
扫描关注微信号,更多精彩内容等着你!
