这是我听B站鲮鱼不会飞视频(R与Bioconductor的入门课)里的笔记哦~
介绍AnnotationHub包
如何下载公共数据并将不同版本的基因组坐标进行互相转换?
这就需要介绍另一个包了叫AnnotationHub
同样,先下载并加载
BiocManager::install("AnnotationHub")
library(AnnotationHub)
载入包之后先给它赋一下值:ah = AnnotationHub()把AnnotationHub包里的数据赋值给一个变量ah。
在赋值的过程中它会自动检查你本地的数据库是不是最新的版本,不是的话会自动更新,更新的过程很慢,耐心等待~
> library(AnnotationHub)
> ah = AnnotationHub()
|===============================================================| 100%
snapshotDate(): 2018-10-24
我们看下AnnotationHub数据库里的数据都是谁来提供的:
table(ah$dataprovider)
从结果中可以看到,这里面常见的数据全都有
我们再看下,它提供了哪些物种的数据呢?
unique(ah$species)
我们从结果中可以看到总共提供了一千多个物种的基因组信息
看下,都提供了哪类的信息:
table(ah$rdataclass)
> table(ah$rdataclass)
AAStringSet BigWigFile
1 10247
biopax ChainFile
9 1113
data.frame data.frame, DNAStringSet, GRanges
40 3
EnsDb FaFile
678 5122
GRanges igraph
20475 1
Inparanoid8Db list
268 18
MSnSet mzRident
1 1
mzRpwiz OrgDb
1 2002
Rle SQLiteConnection
1852 1
TwoBitFile TxDb
5558 75
VcfFile
8
来介绍下:
BigWigFile这个一般是基因组coverage的情况,比如我测了chip-seq,基因组的每一个位置有多少reads能cover住信号值,一般储存为BigWig文件;
ChainFile 是基因组进行坐标转换的。比如,我们人的参考基因组有hg 18/19/38,它们之间坐标互相转换用的就是ChainFile;
FaFile 就是fa文件;
OrgDb我们在做GEO分析的时候用到这个;
TxDb 就是我们的基因组注释。
下面我们学习下,做一个有用的筛选
筛选同时满足这些条件的:K562细胞系,物种是人的chip的数据,它的peaks信息,且数据必须来源于EpigenomeRoadMap组织,代码:
# get more filter info
bpChipEpi <- query(ah , c("EpigenomeRoadMap", "broadPeak", "chip","Homo Sapiens","K562"))
bpChipEpi
# use browser to select data
display(bpChipEpi)
以下就是筛选出来的peaks
> bpChipEpi <- query(ah , c("EpigenomeRoadMap", "broadPeak", "chip","Homo Sapiens","K562"))
> bpChipEpi
AnnotationHub with 12 records
# snapshotDate(): 2018-10-24
# $dataprovider: BroadInstitute
# $species: Homo sapiens
# $rdataclass: GRanges
# additional mcols(): taxonomyid, genome, description,
# coordinate_1_based, maintainer, rdatadateadded, preparerclass,
# tags, rdatapath, sourceurl, sourcetype
# retrieve records with, e.g., 'object[["AH29781"]]'
title
AH29781 | E123-H2A.Z.broadPeak.gz
AH29782 | E123-H3K4me1.broadPeak.gz
AH29783 | E123-H3K4me2.broadPeak.gz
AH29784 | E123-H3K4me3.broadPeak.gz
AH29785 | E123-H3K9ac.broadPeak.gz
... ...
AH29788 | E123-H3K27ac.broadPeak.gz
AH29789 | E123-H3K27me3.broadPeak.gz
AH29790 | E123-H3K36me3.broadPeak.gz
AH29791 | E123-H3K79me2.broadPeak.gz
AH29792 | E123-H4K20me1.broadPeak.gz
我们使用 display(bpChipEpi) 后会弹出一个网页
> display(bpChipEpi)
Listening on http://127.0.0.1:4529
这个网页会告诉你应该选择哪个数据
我们把我们想要的数据选出来,点左上方的Return rows to R session
R就会告诉我们,我们选择的是哪几个。
选完我们想要的数据之后,怎么下载这个peaks数据呢?
chip.peak.K562.H3K4me3 = bpChipEpi[["AH29784"]]
AH29784是数据对应的编号
我们可以看下下载好的chip的数据,长什么样子
> chip.peak.K562.H3K4me3
GRanges object with 100238 ranges and 5 metadata columns:
seqnames ranges strand | name
|
[1] chr9 134000008-134002532 * | Rank_1
[2] chr22 19464765-19468883 * | Rank_2
[3] chr22 19839656-19844037 * | Rank_3
[4] chr22 22335550-22337631 * | Rank_4
[5] chr22 21354340-21358340 * | Rank_5
... ... ... ... . ...
[100234] chr22 19326234-19326924 * | Rank_100234
[100235] chr17 44201260-44201780 * | Rank_100235
[100236] chr1 182168332-182168501 * | Rank_100236
[100237] chr1 233798301-233798556 * | Rank_100237
[100238] chr11 16623577-16623899 * | Rank_100238
score signalValue pValue qValue
[1] 862 8.12246 90.55478 86.25152
[2] 668 10.69822 70.5211 66.85632
[3] 642 10.61566 67.81337 64.21117
[4] 630 8.93212 66.6233 63.07093
[5] 568 8.53532 60.23624 56.88031
... ... ... ... ...
[100234] 0 1.48825 1.00501 0
[100235] 0 1.54538 1.00475 0
[100236] 0 1.54625 1.00167 0
[100237] 0 1.54562 1.00089 0
[100238] 0 1.54562 1.00089 0
-------
seqinfo: 93 sequences (1 circular) from hg19 genome
介绍BioStrings包
如何使用BioStrings处理序列信息?
第一步,永远的下载和加载
biocLite("BioStrings")
library(Biostrings)
我们创建一个DNAstr
str1 = DNAString(x= "ACTGACTGAATTCCGGAAAAAAAA")
> str1
24-letter "DNAString" instance
seq: ACTGACTGAATTCCGGAAAAAAAA
我们可以看到,str1已经是一个DNAString了
我想获得反向序列怎么获得?
reverse(str1)
> reverse(str1)
24-letter "DNAString" instance
seq: AAAAAAAAGGCCTTAAGTCAGTCA
得到反向互补序列:reverseComplement(str1)
> reverseComplement(str1)
24-letter "DNAString" instance
seq: TTTTTTTTCCGGAATTCAGTCAGT
提取DNAString的部分序列:str1[1:10] 前10个序列
> str1[1:10]
10-letter "DNAString" instance
seq: ACTGACTGAA
统计它的长度: length(str1)
假如说我这段序列是一个CDS的序列,我想把它按照标准的密码子表翻译成蛋白的序列,要怎么做呢?
translate(str1) 这个函数就可以办到
> translate(str1)
8-letter "AAString" instance
seq: TD*IPEKK
T-丝氨酸;D-天冬氨酸;*-stopcode;I-异亮氨酸......
统计DNAString中每一个碱基出现了多少次:alphabetFrequency(str1)
> alphabetFrequency(str1)
A C G T M R W S Y K V H D B N - + .
12 4 4 4 0 0 0 0 0 0 0 0 0 0 0 0 0 0
A C G T 我们都知道是啥,那后面的那些是什么?是简并碱基
国际标准的简并碱基是:IUPAC_CODE_MAP
> IUPAC_CODE_MAP
A C G T M R W S Y
"A" "C" "G" "T" "AC" "AG" "AT" "CG" "CT"
K V H D B N
"GT" "ACG" "ACT" "AGT" "CGT" "ACGT"
怎样统计GC含量呢?letterFrequency(str1,"GC")等价于letterFrequency(str1,letters="GC")
计算G/C的比例:letterFrequency(str1,as.prob = T,letters="GC")
> letterFrequency(str1,letters="GC")
G|C
8
> letterFrequency(str1,"GC") #统计GC含量,一共有8个
G|C
8
> letterFrequency(str1,as.prob = T,letters="GC") #计算GC的比例
G|C
0.3333333
> letterFrequency(str1,"GC",as.prob = T)
G|C
0.3333333
我想知道A C G T 这四个里,任意两两的含量的letterFrequency怎么做?
dinucleotideFrequency(str1,as.prob = T)
> dinucleotideFrequency(str1,as.prob = T)
AA AC AG AT CA CC
0.34782609 0.08695652 0.00000000 0.04347826 0.00000000 0.04347826
CG CT GA GC GG GT
0.04347826 0.08695652 0.13043478 0.00000000 0.04347826 0.00000000
TA TC TG TT
0.00000000 0.04347826 0.08695652 0.04347826
A C G T 任意3个字母的含量,也有函数可以直接解决,三个字母的也很有用,因为我们的code都是3个的嘛~
trinucleotideFrequency(str1,as.prob = T)
> trinucleotideFrequency(str1,as.prob = T)
AAA AAC AAG AAT ACA ACC
0.27272727 0.00000000 0.00000000 0.04545455 0.00000000 0.00000000
ACG ACT AGA AGC AGG AGT
0.00000000 0.09090909 0.00000000 0.00000000 0.00000000 0.00000000
ATA ATC ATG ATT CAA CAC
0.00000000 0.00000000 0.00000000 0.04545455 0.00000000 0.00000000
CAG CAT CCA CCC CCG CCT
0.00000000 0.00000000 0.00000000 0.00000000 0.04545455 0.00000000
CGA CGC CGG CGT CTA CTC
0.00000000 0.00000000 0.04545455 0.00000000 0.00000000 0.00000000
CTG CTT GAA GAC GAG GAT
0.09090909 0.00000000 0.09090909 0.04545455 0.00000000 0.00000000
GCA GCC GCG GCT GGA GGC
0.00000000 0.00000000 0.00000000 0.00000000 0.04545455 0.00000000
GGG GGT GTA GTC GTG GTT
0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000
TAA TAC TAG TAT TCA TCC
0.00000000 0.00000000 0.00000000 0.00000000 0.00000000 0.04545455
TCG TCT TGA TGC TGG TGT
0.00000000 0.00000000 0.09090909 0.00000000 0.00000000 0.00000000
TTA TTC TTG TTT
0.00000000 0.04545455 0.00000000 0.00000000
这个包主要是帮我们统计处理一些reverse compliment,是很方便的一些小的工具。
BioStrings这个包里更重要的一个功能是,可以进行双序列比对和多序列比对
但是在R里进行双序列比对和多序列比对不怎么能用的到,所以我们就先不介绍。
介绍BSgenome包
如何获得任意一段human genome 的序列信息?如何统计human各条染色体的长度及GC含量?
需要用到这里要介绍的包BSgenome
BiocManager::install("BSgenome")
library(BSgenome)
BSgenome为我们提供一个标准的注释信息,主要是一些模式生物。
使用命令函数 available.genomes() 可以看到所有已经提供了的基因组。
插曲:基因组下载下来总共能看到有3种序列信息,第一种写的是大写的N,N是没有测出来;除此之外第二种就是大写字母,大写字母表示基因序列复杂度比较高的序列;第三种是小写字母,表示测出来了,复杂度比较低。标记的方法叫softmask
BiocManager::install("BSgenome.Hsapiens.UCSC.hg19")
library(BSgenome.Hsapiens.UCSC.hg19) ##载入hg19的基因组注释信息
hg19.genome = BSgenome.Hsapiens.UCSC.hg19 ##赋值方便我们操作
首先,先看一下我们hg19.genome的序列信息都有哪些~
> hg19.genome
Human genome:
# organism: Homo sapiens (Human)
# provider: UCSC
# provider version: hg19
# release date: Feb. 2009
# release name: Genome Reference Consortium GRCh37
# 93 sequences:
# chr1 chr2 chr3 chr4 chr5
# chr6 chr7 chr8 chr9 chr10
# chr11 chr12 chr13 chr14 chr15
# chr16 chr17 chr18 chr19 chr20
# chr21 chr22 chrX chrY chrM
# ... ... ... ... ...
# chrUn_gl000227 chrUn_gl000228 chrUn_gl000229 chrUn_gl000230 chrUn_gl000231
# chrUn_gl000232 chrUn_gl000233 chrUn_gl000234 chrUn_gl000235 chrUn_gl000236
# chrUn_gl000237 chrUn_gl000238 chrUn_gl000239 chrUn_gl000240 chrUn_gl000241
# chrUn_gl000242 chrUn_gl000243 chrUn_gl000244 chrUn_gl000245 chrUn_gl000246
# chrUn_gl000247 chrUn_gl000248 chrUn_gl000249
# (use 'seqnames()' to see all the sequence names, use the '$' or '[[' operator to access a given sequence)
我们能看到,这里面总共有93个序列(93 sequences)或者说是类基因组的结构。除了1-22号染色体和X/Y染色体之外,还有线粒体染色体,还包括一些我们能测出来但是拼不回去的区域,比如 chr1_gl000191_random 能测到、也知道它是1号染色体就是拼不回去。像那种写着Un的,就是能测出来确定是人的序列,但是拼不回去。
这93个序列怎么查呢?seqnames(hg19.genome)
获得各条染色体的长度: seqlengths(hg19.genome)
我们的基因组从1号染色体到22号染色体,这个顺序大概是按照什么排的?
按照染色体长度从大到小排的。
我们简单的用一下ggplot2画一下这个图,看到底是不是按照从大到小排的。
首先,我们把染色体的长度序列信息组成一个data.frame()叫做hg19.length.df
hg19.length.df = data.frame(chr_name = seqnames(hg19.genome),
chr_len = as.numeric(seqlengths(hg19.genome)))
这就生成了一个两列93行的数据框
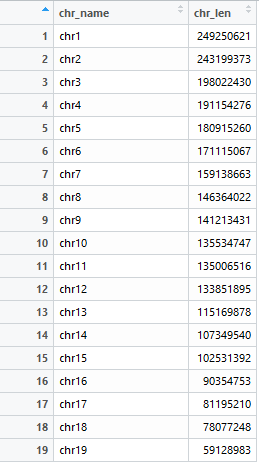
我们这里只想要画前面24条染色体,后面的过滤掉:
hg19.length.df.select = hg19.length.df[1:24,]
View(hg19.length.df.select)
指定下画图的顺序,就是保证最后画出来的图,从1-22号再到X/Y这样画。
hg19.length.df.select$chr_name <- factor(hg19.length.df.select$chr_name,levels = as.character(hg19.length.df.select$chr_name))
用ggplot2作图:
library(ggplot2)
ggplot(hg19.length.df.select,aes(x=chr_name,y=chr_len)) + geom_bar(stat="identity")
ggplot2作图代码解释:首先先给ggplot一个data,再给定X/Y轴,需要画成柱状图所以要加geom_bar参数,
按照它原始的数值,所以给定stat="identity"
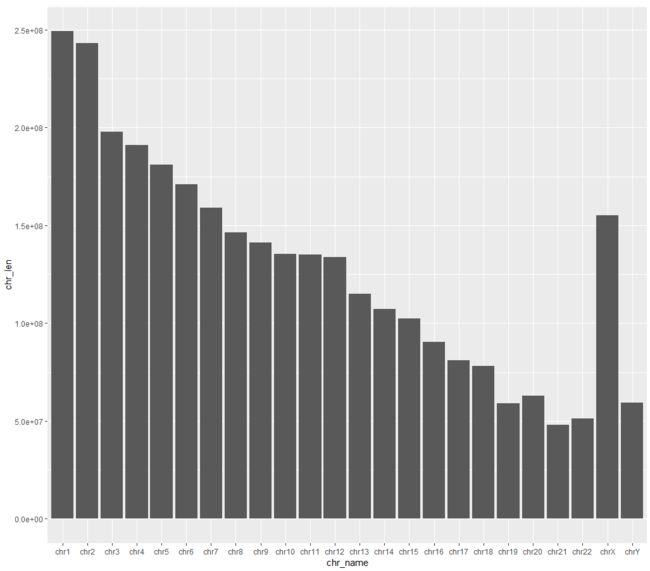
我们从图中可以看到,染色体大概是从大到小排列的,但是我们必须注意一点是人最小的染色体是21号染色体,不是22号(图中能看出来),这是因为在显微镜下21、22这两条染色体基本上是分不出来大小的,先发现的21后发现的22,所以在鉴定的时候就排错了。
回到刚刚的问题:怎么取其中的任意一段序列?
我们已经有了
hg19.genome,我们想取1号染色体上第10万到11万上的序列:
str2 = hg19.genome[["chr1"]][100000:110000]
> str2
10001-letter "DNAString" instance
seq: CACTAAGCACACAGAGAATAATGTCTAGAATCTGAGTGCCATGT...TCCTGTATTAGTCTCCCCAGTAGTTGGGATTACTAGCATATGCC
任意取出一段序列后,怎样统计它的GC含量?和前面一样的
letterFrequency(hg19.genome[["chr1"]],"GC",as.prob = T)
> letterFrequency(hg19.genome[["chr1"]],"GC",as.prob = T)
G|C
0.3772948
也就是说人的1号染色体的GC含量大概是37%左右
如何使用 rtracklayer 读取文件(以GTF文件为例)
rtracklayer是专门读取文件的,它可以读非常多的文件类型,比如:BED / bigwig / GFF / GTF 还有其他的一些coverage的信号的文件,一般比较常见的格式文件他都可以读进R来。
BiocManager::install("rtracklayer")
library(rtracklayer)
gtf文件直接读进来就变成我们上面重点讲过的GRanges。这次我们读进来一个hg19的 refseq 基因:
hg19.RefSeq.gtf = import.gff(con="~/文件所在路径/")
这样就读完了
看下它有多长
length(hg19.RefSeq.gtf)
gtf文件主要显示以下信息:前面显示几号染色体;给一个位置区间;告诉你正负链;数据来源;是exon还是CDS;告你基因ID;转录本的ID
读进来就是一个GRanges,具体怎么操作,向上翻翻有很详细的介绍的。
如何从UCSC genome browser下载GTF/GFF文件
- 打开UCSC genome browser网站
- 在Tools里选择Table Browser
- 打开Table Browser以后,设置相关的需要内容(如下图)
-
点击get output即可下载
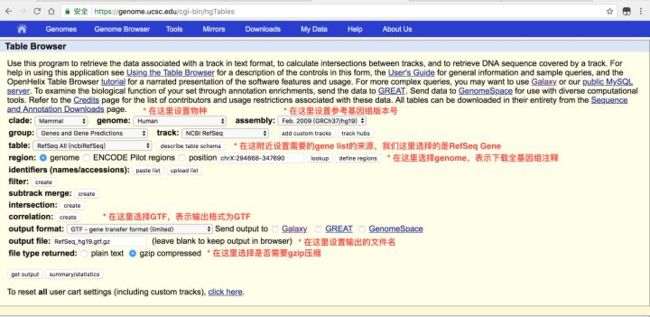 Table Browser如何设置
Table Browser如何设置
Tips:hg19 = human genome 19是常用的human参考基因组版本号;RefSeq gene是全部经过人工检查过的gene注释文件。
如何将不同版本的基因组坐标相互转换?
这是一个非常重要的问题,我们举个例子,我们平时实战的时候经常会遇到这样一个问题:我们有一段区域,比对到的参考基因组是hg19,但是另一个公共数据人家比对到的是hg38,为了让这两个之间有可比性,那我们就需要将这段区域在hg19的位置映射到hg38,在R里非常方便,需要用到chain file 这个需要用AnnotationHub下载,AnnotationHub的包我们已经讲过了
library(AnnotationHub)
ah <- AnnotationHub()
载入这个包之后给它赋值一下。
我们先看下ah里面提供了哪些数据类型:table(ah$rdataclass)
> table(ah$rdataclass)
AAStringSet BigWigFile
1 10247
biopax ChainFile
9 1113
data.frame data.frame, DNAStringSet, GRanges
40 3
EnsDb FaFile
678 5122
GRanges igraph
20475 1
Inparanoid8Db list
268 18
MSnSet mzRident
1 1
mzRpwiz OrgDb
1 2002
Rle SQLiteConnection
1852 1
TwoBitFile TxDb
5558 75
VcfFile
8
这里面FaFile就是提供基因组的序列信息;ChainFile提供坐标转换,提供了1113 种坐标转换的方式。
我们尝试着进行hg19和hg38之间的转换
首先我们进行下搜索(query),在ah下搜索,搜索的关键词是"hg38", "hg19", "chainfile"
chainfiles <- query(ah , c("hg38", "hg19", "chainfile"))
搜索完之后发现,能搜索到hg19到hg38也能搜索到hg38到hg9
> chainfiles
AnnotationHub with 2 records
# snapshotDate(): 2018-10-24
# $dataprovider: UCSC
# $species: Homo sapiens
# $rdataclass: ChainFile
# additional mcols(): taxonomyid, genome, description,
# coordinate_1_based, maintainer, rdatadateadded,
# preparerclass, tags, rdatapath, sourceurl, sourcetype
# retrieve records with, e.g., 'object[["AH14108"]]'
title
AH14108 | hg38ToHg19.over.chain.gz
AH14150 | hg19ToHg38.over.chain.gz
我们把其中的一个文件下载下来,并命名为hg19.to.hg38.chain:
hg19.to.hg38.chain = chainfiles[["AH14150"]]
> hg19.to.hg38.chain = chainfiles[["AH14150"]]
downloading 1 resources
retrieving 1 resource
|===========================================================| 100%
loading from cache
‘C:/Users/huangjing00liang/Documents/AppData/.AnnotationHub/18245’
hg19 转成 hg38
> test.GR = head(hg19.RefSeq.gtf) #就用这个gtf文件的前几行做示范
> test.GR.hg38 = liftOver(test.GR, hg19.to.hg38.chain)
### liftOver()后面先输入原来的那个坐标文件(这里就是test.GR),再给`chainfiles`就可以了
> unlist(test.GR.hg38) #让输出文件变得正常一些
如果你有一个SNP位点是hg19上的,你想让它变成hg38上,就需要经过上述处理。
如何使用 GEOquery获得GEO数据库里面的数据?
同样,下载并载入GEOquery
BiocManager::install("GEOquery")
library(GEOquery)
一篇好文章在GEO数据库会有好多好多文件,我们不可能一个一个去下载,我们这里的GEOquery就可以批量下载全部文件。
用GEO编号,来下载数据
# get list from GEO
eList = getGEO("GSE36552")
最后会得到数据存放位置,说明下载成功了
File stored at:
C:\Users\HUANGJ~1\AppData\Local\Temp\Rtmpygkwdf/GPL11154.soft
用length(eList)看下它的长度,发现是1,其实是因为现在它还是一个列表,我们需要把列表里面的数据提取出来:eData = eList[[1]] 这个时候会得到一个ExpressionSet 这个后续进阶我们一定会好好讲,现在会用就可以了。
> length(eList)
[1] 1
> eData = eList[[1]]
> eData
ExpressionSet (storageMode: lockedEnvironment)
assayData: 0 features, 124 samples
element names: exprs
protocolData: none
phenoData
sampleNames: GSM896803 GSM896804 ... GSM922275 (124 total)
varLabels: title geo_accession ... tissue:ch1 (45 total)
varMetadata: labelDescription
featureData: none
experimentData: use 'experimentData(object)'
pubMedIds: 23934149
Annotation: GPL11154
但凡看到写着ExpressionSet,就会有以下几个函数可以用:
- pData(eData)
该函数返回的是,该矩阵所有的phenodata(表型相关的数据)或者叫做metadata - sampleNames(eData)
返回结果如下
> sampleNames(eData)
[1] "GSM896803" "GSM896804" "GSM896805" "GSM896806" "GSM896807"
[6] "GSM896808" "GSM896809" "GSM896810" "GSM896811" "GSM896812"
[11] "GSM896813" "GSM896814" "GSM896815" "GSM896816" "GSM922146"
[16] "GSM922147" "GSM922148" "GSM922149" "GSM922150" "GSM922151"
[21] "GSM922152" "GSM922153" "GSM922154" "GSM922155" "GSM922156"
[26] "GSM922157" "GSM922158" "GSM922159" "GSM922160" "GSM922161"
[31] "GSM922162" "GSM922163" "GSM922164" "GSM922165" "GSM922166"
[36] "GSM922167" "GSM922168" "GSM922169" "GSM922170" "GSM922171"
[41] "GSM922172" "GSM922173" "GSM922174" "GSM922175" "GSM922176"
[46] "GSM922177" "GSM922178" "GSM922179" "GSM922180" "GSM922181"
[51] "GSM922182" "GSM922183" "GSM922184" "GSM922185" "GSM922186"
[56] "GSM922187" "GSM922188" "GSM922189" "GSM922190" "GSM922191"
[61] "GSM922192" "GSM922193" "GSM922194" "GSM922195" "GSM922196"
[66] "GSM922197" "GSM922198" "GSM922199" "GSM922200" "GSM922201"
[71] "GSM922202" "GSM922203" "GSM922204" "GSM922205" "GSM922206"
[76] "GSM922207" "GSM922208" "GSM922209" "GSM922210" "GSM922211"
[81] "GSM922212" "GSM922213" "GSM922214" "GSM922215" "GSM922216"
[86] "GSM922217" "GSM922218" "GSM922219" "GSM922220" "GSM922221"
[91] "GSM922222" "GSM922223" "GSM922224" "GSM922225" "GSM922226"
[96] "GSM922227" "GSM922228" "GSM922230" "GSM922250" "GSM922251"
[101] "GSM922252" "GSM922253" "GSM922254" "GSM922255" "GSM922256"
[106] "GSM922257" "GSM922258" "GSM922259" "GSM922260" "GSM922261"
[111] "GSM922262" "GSM922263" "GSM922264" "GSM922265" "GSM922266"
[116] "GSM922267" "GSM922268" "GSM922269" "GSM922270" "GSM922271"
[121] "GSM922272" "GSM922273" "GSM922274" "GSM922275"
pheno.df = pData(eData)
赋完值之后我们看下这个表,最左边的是对应的GEO编号;title是来自于哪个细胞,如图里的Oocyte就是来自于卵母细胞,Zygote就是合子;再往后就是一些具体的信息,比如taxid_chr1那列9606就表示人的分类学ID,9606就代表人,所以说它的来源是人。后面的列还会告诉你它用了什么样的操作,relation那列比较重要,它会告诉你SRA原始文件(也就是fa文件)的网址。
我们这里想要得到的表达量信息在supplementary_file_2那列,该列放着这篇文章100多个样本的数据信息网址链接。链接找到了离成功批量下载数据就不远了。我们把这些链接单独存在一个文件里,然后去服务器上wget一下就全下载下来了。
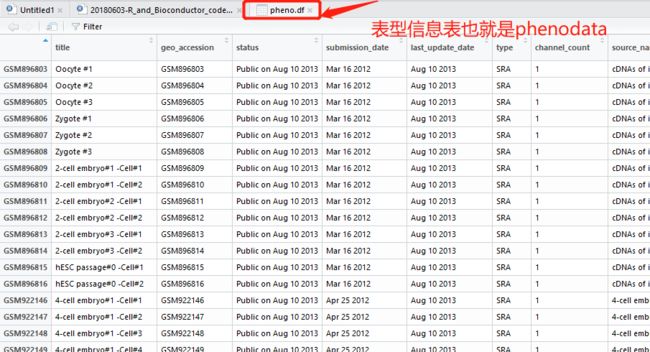
如何把我们想要的存放样本网址的这一列提取出来呢?这好办~
用
$把
pheno.df数据框中想要的那一列取出来然后赋值给一个新的变量
html.df
> html.df = pheno.df$supplementary_file_2
> html.df
[1] ftp://ftp.ncbi.nlm.nih.gov/geo/samples/GSM896nnn/GSM896803/suppl/GSM896803_Sample_M21.expression.txt.gz
[2] ftp://ftp.ncbi.nlm.nih.gov/geo/samples/GSM896nnn/GSM896804/suppl/GSM896804_Sample_M22.expression.txt.gz
[3] ftp://ftp.ncbi.nlm.nih.gov/geo/samples/GSM896nnn/GSM896805/suppl/GSM896805_Sample_M23.expression.txt.gz
[4] ftp://ftp.ncbi.nlm.nih.gov/geo/samples/GSM896nnn/GSM896806/suppl/GSM896806_Sample_C1.expression.txt.gz
取出网址这一列后,我们把它储存一下:
> html.df = as.data.frame(html.df )
> View(html.df)
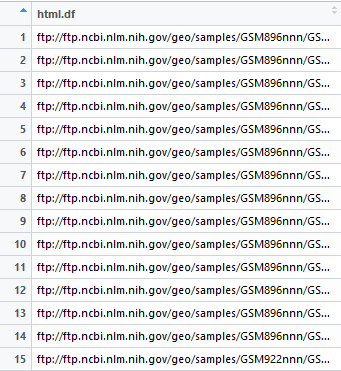
再把这个文件写一下也就是把它存到本地:
> write.csv(html.df,file = "~/test.txt",row.names = F,quote = F)
本地就多了一个test.txt文档了

拿这个文本文件去服务器上
wget就可以批量下载啦~
我之前都是直接去GEO数据库点开链接就直接下载到电脑上了,下载下来是一个压缩包,然后传到服务器上,这种操作样本量少的时候是可以的,如果样本量特别大就不行了,一是占电脑太多内存,二是数据量太大的话从电脑传到服务器上也耗时呀!
介绍Gviz包
如何将公共数据的chip-seq peak信息与基因结构信息进行批量可视化?
我们前面已经讲过怎么样用AnnotationHub下载chipseq的公共数据,现在我们进行批量可视化。
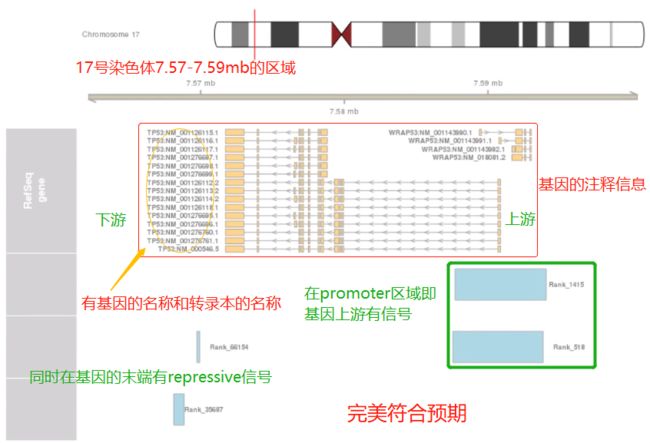
需要用到Gviz这个包,需要到达的目的是:

上图每一行其实是不同的Track:
- 第一行那个黑白相间的横条叫
ideogram track - 第二行是
chromosome track - 第三行是
data info track - 第四行是
gene annotation track
我们先来设计下我们的问题:
- 第1行画
ideogram track - 第2行画
tick track / aixs track - 第3行画
gene annotation track - 第4—6行画
chip-seq track
注:我们chipseq选三个分别是:两个active的信号和一个repressive的信号
总共我们需要有6个track
画图的思路大概分成四个部分:假如我们有一个基因是TP53(gene_symbol = "TP53")
- 第一步应该把gtf文件里面等于
gene_symbol的取出来了,也就是把有关TP53基因的所有信息提取出来,获得TP53基因的起始位置和终止位置; - 第二步把起始位置和终止位置适当的延长,比如我们延长
10K或者5K都无所谓,取出延长后这段的所有基因; - 第三步有了起始终止位置就可以画
ideogram track和aixs track; - 第四步对chip-seq数据进行select,画出该区域的
chip-seq track。
前方代码有些长,做好准备,跟紧啦~~~
rm(list=ls()) 好习惯要养成,先把所有的东西清空一下
先用AnnotationHub下载要用的chip-seq的公共数据
ah = AnnotationHub()
然后进行过滤,得到过滤之后的信息
bpChipEpi <- query(ah , c("EpigenomeRoadMap", "broadPeak", "chip","Homo Sapiens","K562"))
bpChipEpi
> bpChipEpi #得到12条注释信息
AnnotationHub with 12 records
title
AH29781 | E123-H2A.Z.broadPeak.gz
AH29782 | E123-H3K4me1.broadPeak.gz
AH29783 | E123-H3K4me2.broadPeak.gz
AH29784 | E123-H3K4me3.broadPeak.gz
AH29785 | E123-H3K9ac.broadPeak.gz
... ...
AH29788 | E123-H3K27ac.broadPeak.gz
AH29789 | E123-H3K27me3.broadPeak.gz
AH29790 | E123-H3K36me3.broadPeak.gz
AH29791 | E123-H3K79me2.broadPeak.gz
AH29792 | E123-H4K20me1.broadPeak.gz
如前所述,我们选3个chipseq的信号——H3K4me3,H3K27ac,H3K27me3
运行以下代码就可以把这3个peaks进行下载。
我们从上面看到H3K4me3 的 active signal 的title是AH29784
chip.peak.K562.H3K4me3 = bpChipEpi[["AH29784"]]
我们从上面看到H3K27ac的 active singal 的title是AH29788
chip.peak.K562.H3K27ac = bpChipEpi[["AH29788"]]
我们从上面看到H3K27me3的 repressive singal 的title是AH29789
chip.peak.K562.H3K27me3 = bpChipEpi[["AH29789"]]
上面那三个peaks信号下载的很慢,不知道是不是网速的问题
下载好之后,先拿一个简单看下:
head(chip.peak.K562.H3K4me3)
chip的peak都是不分正负链的,我看下score的分布:
hist(chip.peak.K562.H3K4me3$score)
发现绝大多数的score都是很低的,有少部分比较高,我们画peak的话,score必须大于0,所以我们先对这三个进行筛选(score > 0)
chip.peak.K562.H3K27me3 = chip.peak.K562.H3K27me3[chip.peak.K562.H3K27me3$score > 0]
chip.peak.K562.H3K27ac = chip.peak.K562.H3K27ac[chip.peak.K562.H3K27ac$score > 0]
chip.peak.K562.H3K27me3 = chip.peak.K562.H3K27me3[chip.peak.K562.H3K27me3$score > 0]
不这样筛选(score > 0)一下的话,会出现一些比较杂的信号
下一步,用Refseq加载gtf文件
hg19.RefSeq.gtf = import.gff(con="~/文件所在路径/")
画图的时候有一个需求:在最后的图上不但要展示基因名还要展示转录本名,因此,我们需要把基因名和转录本名拼出来。同时Gviz在画图的时候必须得把gene_id改成基因名 transcript_id改成transcript,也就是不能有_id,因此我们重新创建一个,用我们之前讲过的GRanges
hg19.RefSeq.gtf.new = GRanges(hg19.RefSeq.gtf,
source = hg19.RefSeq.gtf$source,
type = hg19.RefSeq.gtf$type,
score = hg19.RefSeq.gtf$score,
phase = hg19.RefSeq.gtf$phase,
gene = hg19.RefSeq.gtf$gene_id,
transcript = hg19.RefSeq.gtf$transcript_id,
symbol = sprintf("%s:%s",hg19.RefSeq.gtf$gene_id,hg19.RefSeq.gtf$transcript_id))
上面的代码意思大概就是,我要创建一个新的hg19的gtf文件(满足我们作图要求的),用GRanges还是在原来gtf文件(hg19.RefSeq.gtf)基础上,source、 type 、 score 、 phase都和原来文件中的一样(source = hg19.RefSeq.gtf$source)。
下面就需要注意一下了:
我们把gene_id改成 gene了(gene = hg19.RefSeq.gtf$gene_id);
transcript_id改成了transcript了(transcript = hg19.RefSeq.gtf$transcript_id);
最后这个命令也是理解一下:
symbol = sprintf("%s:%s",hg19.RefSeq.gtf$gene_id,hg19.RefSeq.gtf$transcript_id)
我们为symbol重新赋值了,怎么赋值的呢?%s表示字符串,%s:%s前面的字符串赋值为hg19.RefSeq.gtf$gene_id 后面的字符串赋值为hg19.RefSeq.gtf$transcript_id
我们看一下新生成的hg19.RefSeq.gtf.new:head(hg19.RefSeq.gtf.new)
这个文件我们准备好了,我们先画一个基因的图
gene_symbol = "TP53"
选出我们想要的信息(select refseq)
RefSeq.gtf.select = hg19.RefSeq.gtf.new[hg19.RefSeq.gtf.new$gene == gene_symbol] # 先把所有名字叫TP53的基因选出来
region.start = min(start(RefSeq.gtf.select)) - 10000 #基因序列开始的位置
region.end = max(end(RefSeq.gtf.select)) + 10000 #基因序列结束的位置
region.chr_name = as.character(seqnames(RefSeq.gtf.select))[1]
#下面我们要把这个区域里面所有和这个区域有关的基因全部都选择出来
select_region.GR_obj = GRanges(seqnames = region.chr_name,
ranges = IRanges(start = region.start,end = region.end))
select_region.GR_obj
#这样我们就把增加过长度的区域也做成了`GRanges`
我们要从最原始的hg19.RefSeq.gtf.new文件里面,筛选出来我们最后用来画图的所有的基因region
# select region all genes
region.Refseq.select = hg19.RefSeq.gtf.new[hg19.RefSeq.gtf.new %over% select_region.GR_obj]
此时,就筛选完成 select region all genes
我们可以用Gviz画第一部分了:ideogram
BiocManager::install("Gviz")
library(Gviz)
ideogram_track = IdeogramTrack(chromosome = region.chr_name,genome = "hg19")
第二部分:axis track
axis_track = GenomeAxisTrack(range = IRanges(start = region.start,end = region.end),genome="hg19")
第三部分:gene annotation
gene_track = GeneRegionTrack(region.Refseq.select,
genome = "hg19",
chromosome = region.chr_name,
name = "ReSeq gene",
transcriptAnnotation = "symbol")
第四—第六部分:chip-seq
第4部分:H3K4me3
chip.peak.K562.H3K4me3.select = chip.peak.K562.H3K4me3[chip.peak.K562.H3K4me3 %over% select_region.GR_obj]
chip.H3K4me3.track = AnnotationTrack(start = start(chip.peak.K562.H3K4me3.select),
width = width(chip.peak.K562.H3K4me3.select),
strand = strand(chip.peak.K562.H3K4me3.select),
genome = "hg19",
chromosome = region.chr_name,
name = "K562.H3K4me3",
group = as.character(chip.peak.K562.H3K4me3.select$name),
groupAnnotation = "group",
just.group = "right"
)
#这里需要注意一下,group就是每一个peak的名字,所以我们要选一个unique的名字,这里unique的名字就是`$name`那列,我们按照group的方式进行注释( groupAnnotation = "group")且把group名称放到右面(just.group = "right")。
第5部分:H3K27ac 只是把上面的代码中H3K4me3换成H3K27ac即可
chip.peak.K562.H3K27ac.select = chip.peak.K562.H3K27ac[chip.peak.K562.H3K27ac %over% select_region.GR_obj]
chip.H3K27ac.track = AnnotationTrack(start = start(chip.peak.K562.H3K27ac.select),
width = width(chip.peak.K562.H3K27ac.select),
strand = strand(chip.peak.K562.H3K27ac.select),
genome = "hg19",
chromosome = region.chr_name,
name = "K562.H3K27ac",
group = as.character(chip.peak.K562.H3K27ac.select$name),
groupAnnotation = "group",
just.group = "right"
)
第6部分:H3K27me3 只是把上面的代码中H3K27ac换成H3K27me3即可
chip.peak.K562.H3K27me3.select = chip.peak.K562.H3K27me3[chip.peak.K562.H3K27me3 %over% select_region.GR_obj]
chip.H3K27me3.track = AnnotationTrack(start = start(chip.peak.K562.H3K27me3.select),
width = width(chip.peak.K562.H3K27me3.select),
strand = strand(chip.peak.K562.H3K27me3.select),
genome = "hg19",
chromosome = region.chr_name,
name = "K562.H3K27me3",
group = as.character(chip.peak.K562.H3K27me3.select$name),
groupAnnotation = "group",
just.group = "right"
)
所以到目前为止我们6个track构建好了,开始画图,画图的时候需要把这些track存到一个list里面
plot_track_list = list(ideogram_track,
axis_track,
gene_track,
chip.H3K4me3.track,
chip.H3K27ac.track,
chip.H3K27me3.track)
#6个track存到一个list里面——plot_track_list
最后一步,plotTracks 作图
plotTracks(plot_track_list, # 先把整合好的list放进去
from = region.start,
to = region.end,
showID = T, # 要不要展示我的ID,T要
sizes = c(1,1,2,1,1,1), #设置每一列的高度
chromosome = region.chr_name,
genome = "hg19")
注意: sizes参数是设置每一列的高度,根据经验,基因的那个地方稍微高一点,其他地方都是1就可以。
所谓批量产图就是,换一下我们这里的唯一变量gene_symbol就行,
把所有的基因名写入到for循环里面,然后把剩下的代码都括住就批量产图了。
参考资料:
https://www.bilibili.com/video/av49363776?from=search&seid=10658324414632164518
https://www.bilibili.com/video/av26731585?from=search&seid=10658324414632164518
https://www.bilibili.com/video/av25643438?from=search&seid=10658324414632164518
https://www.bilibili.com/video/av37568990?from=search&seid=10658324414632164518
https://www.bilibili.com/video/av24355734
https://www.bilibili.com/video/av29255401?from=search&seid=10658324414632164518