https://hbctraining.github.io/DGE_workshop/lessons/01_DGE_setup_and_overview.html
(今天状态不佳,加上统计学基础不怎么样,如有错误请指出,我将在中更新)
学习目标
- 解释实验及其目标
- 描述如何在R中设置RNA-seq项目
- 描述RNA-seq和差异基因表达分析工作流程
- 解释为什么负二项分布用于模拟RNA-seq计数数据
差异基因表达(DGE)分析概述
RNA-seq的目标通常是进行差异表达检测,以确定哪些基因在不同条件间表达量有差异。这些基因可以从生物学角度揭示不同条件下受影响的生物过程。
RNA-seq工作流程的详细步骤如下图,可用于确定基因的表达水平。通过生成每个基因的read counts,在命令行(Linux / Unix)上执行所有步骤。差异表达分析和其他下游功能分析通常用R中的专门的程序包完成,这些R包是为完成差异分析所需的复杂统计分析而设计的。

在接下来的几节课中,我们将引导你使用各种R包进行end-to-end的基因水平RNA-seq差异表达工作流程。我们将从计数矩阵开始,进行质控的探索性数据分析,探索样本之间的关系,进行差异表达分析,并在执行下游功能分析之前直观地探索结果。
1.检查示例数据集
示例数据集是RNA-Seq的完整计数矩阵,是Kenny PJ et al,Cell Rep 2014(http://www.ncbi.nlm.nih.gov/pubmed/25464849)描述的更大研究的一部分。
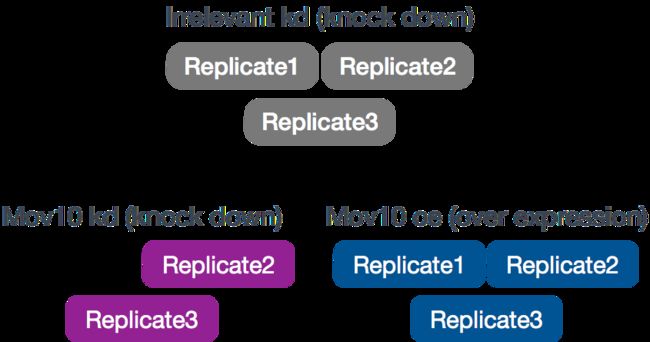
RNA-Seq实验对象是HEK293F细胞,所述HEK293F细胞用MOV10转基因或siRNA转染以降低Mov10表达,或非特异性(无关)siRNA转染。这导致了3种情况:Mov10 oe(过表达),Mov10 kd(击倒)和不相关的kd。重复次数如下所示。
使用这些数据,我们将评估与MOV10表达的扰动相关的转录模式。请注意,不相关的siRNA作为对照组。
这些数据集的目的是什么?Mov10做了什么?
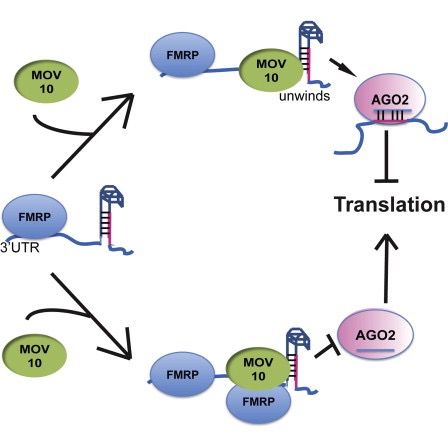
作者正在研究脆性X综合征中涉及的各种基因之间的相互作用,这是一种FMRP蛋白异常产生的疾病。
FMRP “最常见于大脑,对正常的认知发育和女性生殖功能至关重要。该基因的突变可导致脆性X综合征,精神发育迟滞,卵巢早衰,自闭症,帕金森病,发育迟缓和其他认知缺陷。“ - 来自维基百科
MOV10是推定的RNA解旋酶,其在microRNA途径的背景下也与FMRP相关。
该论文正在测试的假说是FMRP和MOV10结合并调节RNA子集的翻译。
问题:
- 可以通过MOV10的丢失或获得来识别哪些表达模式?
- 两种情况之间是否有共同的基因?
2.组织工作目录
在深入了解分析的细节之前,先打开RStudio并为此分析设置一个新项目。
- 转到
File菜单并选择New Project。 - 在
New Project窗口中,选择New Directory。然后,选择Empty Project。为新目录命名DEanalysis,然后将“创建项目作为子目录:”桌面(或自定义的位置)。 - 新的project应该在RStudio中自动打开了。
要检查是否在正确的工作目录中,请使用getwd()。在控制台中应该会返回路径Desktop/DEanalysis。在工作目录下使用New folder按钮创建三个新目录:data,meta和results。请记住,好的分析的关键是从一开始就保持井井有条!
转到File菜单并选择New File,然后选择R Script。此时在Rstudio左上角打开了一个脚本编辑器。这是接下来输入和保存此分析所需的所有命令的地方。在脚本编辑器中输入标题行:
## Gene-level differential expression analysis using DESeq2
现在将文件另存为de_script.R。完成后,工作目录应如下图:
最后,需要获取我们将用于分析的文件。右键单击下面的链接,然后选择“将链接另存为...”选项进行下载:
- 将counts矩阵(https://raw.githubusercontent.com/hbc/NGS_Data_Analysis_Course/master/sessionIII/data/Mov10_full_counts.txt)文件保存在
data目录中。 - 将metadata数据表(https://raw.githubusercontent.com/hbc/NGS_Data_Analysis_Course/master/sessionIII/data/Mov10_full_meta.txt)文件保存在
meta目录中。
4. 加载包
对于这个分析用到的R包,有的是从CRAN安装的,有的是从Bioconductor安装的。要使用这些包(以及它们中包含的函数),我们需要加载包。将以下内容添加到脚本中,可增加自主注释
## Setup
### Bioconductor and CRAN libraries used
library(tidyverse)
library(RColorBrewer)
library(DESeq2)
library(pheatmap)
library(DEGreport)
5. 载入数据
要将数据加载到我们当前的环境中,我们将使用该read.table功能。我们需要提供每个文件的路径,并指定参数让R知道我们有一个header(header = T),第一列是我们的行名(row.names =1)。默认情况下,该函数需要使用制表符分隔的文件,这就是我们所拥有的。
## Load in data
data <- read.table("data/Mov10_full_counts.txt", header=T, row.names=1)
meta <- read.table("meta/Mov10_full_meta.txt", header=T, row.names=1)
使用class()来检查我们的数据:
### Check classes of the data we just brought in
class(meta)
class(data)
6. 查看数据
在继续执行任何类型的分析之前,请确保数据集包含预期的样本/信息。
View(meta)
View(data)
使用Salmon的丰度估计值作为DESeq2的输入
课程中使用的counts是使用RNA-seq分析的标准方法生成的,其中使用剪接感知对准器将样本与基因组比对,然后计数。如果使用轻量级算法(如Salmon,Sailfish或Kallisto)估算丰度,还可以使用DESeq2执行基因水平差异表达分析。这些转录本丰度估计值(通常称为“伪计数”)可以转换为与DESeq2一起使用,但设置稍微复杂一些。有关使用DESeq2的Salmon伪载体的更多信息,详见https://hbctraining.github.io/DGE_workshop_salmon/lessons/01_DGE_setup_and_overview.html。
那么counts数据实际代表什么呢?用于差异表达分析的counts数据表示源自特定基因的序列reads的数量。counts越多,与该基因相关的reads数越多,就假设样本中该基因的表达水平越高。

差异表达分析可以寻找两个或更多组(在meta数据中定义)之间表达量发生变化的基因
- 处理与对照
- 表达与某些变量或临床结果的相关性
为什么不能根据两组之间的差异(基于倍数变化值)对基因进行排序,来识别差异表达的基因?
通常情况下,得到的数据会比预期的要多得多。样品之间表达水平不同的基因不仅是感兴趣的实验变量的结果,而且与一些外界因素有关。差异表达分析的目的是确定这些因素的相对作用,并将“interesting”与“ uninteresting”的因素分开。
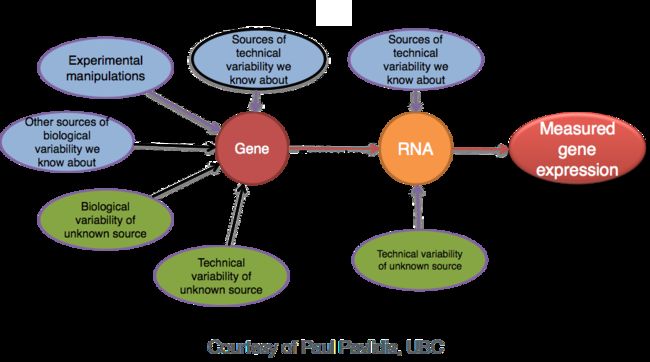
“ uninteresting”的因素也会导致数据的差异,因此即使样本组之间的平均表达水平看起来相差很大,但实际上可能差异并不显著。这是针对下图中“GeneA”的“untreated”组和“treated”之间表达进行说明的。'treated'组的geneA的平均表达水平是'untreated'组的两倍,但重复之间的差异表明,这可能不是显着差异。在确定基因是否差异表达时,需要考虑数据的变化(以及它可能来自何处)。

差异表达分析的目标是针对每个基因确定组间表达(计数)的差异是否显着,给定组内(重复)观察到的变异量。为了测试显著性,我们需要一个适当的统计模型,准确地执行标准化(以解释测序深度的差异等)和方差建模(以考虑少数重复和大动态表达范围)。
RNA-seq计数分布
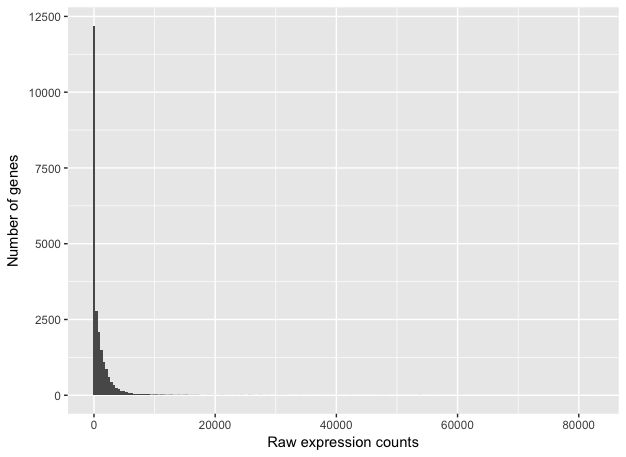
为了确定适当的统计模型,需要有关计数分布的信息。绘制样本'Mov10_oe_1'的计数分布图:
ggplot(data) +
geom_histogram(aes(x = Mov10_oe_1), stat = "bin", bins = 200) +
xlab("Raw expression counts") +
ylab("Number of genes")
如果我们放大到接近零,我们可以看到大量基数为零:
ggplot(data) +
geom_histogram(aes(x = Mov10_oe_1), stat = "bin", bins = 200) +
xlim(-5, 500) +
xlab("Raw expression counts") +
ylab("Number of genes")
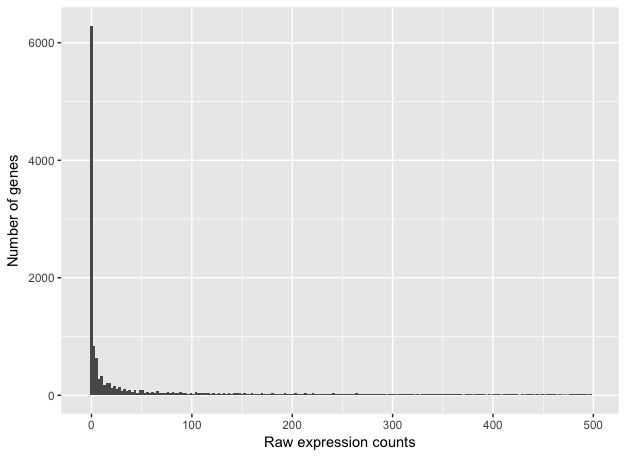
上图说明了RNA-seq计数数据的一些普遍特征:大部分基因计数很少,以及由于表达量低到图上不显示而具有很长的右尾。与芯片数据不同,芯片数据由于探针有最大限度,counts的动态范围有最大值,而RNA-seq数据没有。由于这些技术的不同,RNA-Seq和芯片用于拟合数据的统计模型也是不同的。
注意:芯片数据的对数强度接近正态分布。而由于RNA-seq计数数据的不同性质,例如整数计数而不是连续测量和非正态分布数据,正态分布不能准确地模拟RNA-seq计数[ 1 ]。
7.counts数据建模
计数数据通常使用二项分布建模,这可以让你有可能在投掷硬币多次时获得多个heads(不会翻译)。但是,并非所有计数数据都适合二项分布。二项式基于离散事件,有确定数量的样本时使用。
样本(即购买彩票的人)数量非常大,但事件(获胜的概率)的概率非常小,泊松分布用于模拟这些类型的计数数据。泊松与二项式类似,但基于连续事件。详见Rafael Irizarry在EdX课程(https://youtu.be/fxtB8c3u6l8)
使用RNA-Seq数据,有非常多的RNA,并且抽出特定转录本的概率非常小。因此,使用泊松分布适合这种情况。但是,此分布的唯一属性是均值==方差。实际上,对于RNA-Seq数据,重复中总是存在一些生物学变异(在样本类中)。具有较大平均表达水平的基因倾向于在重复中具有较大的观察到的变异。
如果mRNA的比例在每个样本类别的生物学重复之间保持完全恒定,我们可以预期泊松分布(其中均值==方差)。Rafael Irizarry在EdX课程(https://youtu.be/HK7WKsL3c2w)中对这个概念进行了很好的描述。但这在实践中不会发生,因此泊松分布被认为仅适用于单个生物样本。
考虑到重复之间的这种类型的可变性,最适合的模型是负二项(NB)模型。从本质上讲,NB模型是平均值<方差的数据的良好拟合,就像RNA-Seq计数数据的情况一样。
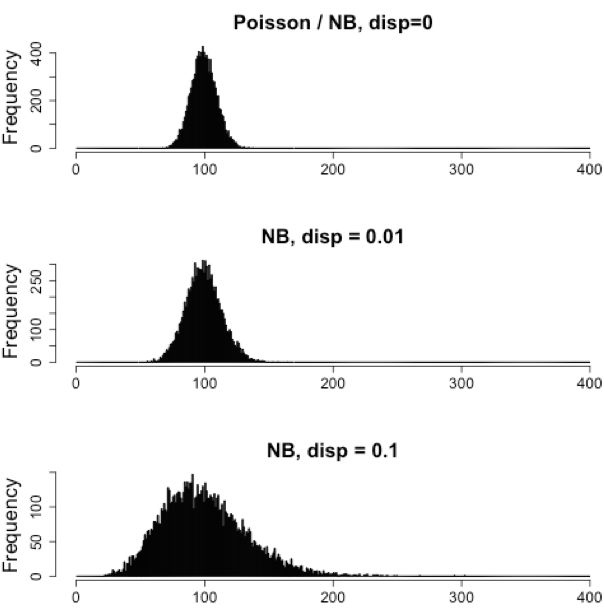
注意:
- 生物学重复代表相同样品类别的多个样品(即来自不同小鼠的RNA)
- 技术重复代表相同的样品(即来自相同小鼠的RNA),但复制了技术步骤
- 通常生物学方差远大于技术方差,因此我们不需要考虑技术方差来识别表达中的生物学差异
- 不要在技术重复上花钱 - 生物复制更有用
注意: 如果使用细胞系并且不确定是否准备了生物学或技术重复,请查看此链接。这是一个有用的资源,可帮助确定如何最好地设置体外实验。
如何知道我的数据是否应使用泊松分布或负二项分布建模?
如果它是计数数据,它应该适合负二项式,如前所述。但是,绘制平均值与数据方差的关系可能会有所帮助。记住泊松模型,均值=方差,但负二项分布的NB均值<方差。
运行以下代码以绘制'Mov10过表达'重复的平均值与方差:
mean_counts <- apply(data[, 3:5], 1, mean)
variance_counts <- apply(data[, 3:5], 1, var)
df <- data.frame(mean_counts, variance_counts)
ggplot(df) +
geom_point(aes(x=mean_counts, y=variance_counts)) +
geom_line(aes(x=mean_counts, y=mean_counts, color="red")) +
scale_y_log10() +
scale_x_log10()

注意,在上图中,重复的变异倾向于大于平均值(红线),特别是对于具有大平均表达水平的基因。这是一个很好的迹象,表明我们的数据不符合泊松分布,我们需要使用负二项模型来解释这种方差的增加(即泊松会低估导致假阳性DE基因增加的变异性)。
通过生物学重复改善平均值估计(即减少方差)
随着生物学重复的数量(随着重复次数的增加,分布将接近泊松分布),方差或散射倾向于减少,因为平均值的标准偏差小于个体观察的标准偏差。额外重复的价值在于,当添加更多数据(重复)时,可以获得更精确的组均值估计,并最终对区分样本类别(即更多DE基因)之间差异的能力更有信心。
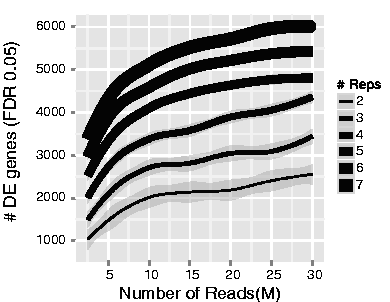
下图说明了测序深度与重复数量之间的关系,鉴定了差异表达基因的数量 (https://academic.oup.com/bioinformatics/article/30/3/301/228651/RNA-seq-differential-expression-studies-more) 。注意,与增加测序深度相比,重复数量的增加倾向于返回更多的DE基因。因此,通常增加重复比增加测序深度更好,但需要注意的是,检测低表达的差异基因和进行同种型水平的差异表达需要更高的测序深度。一般来说,建议的最小测序深度是每个样品20-30万个reads,但如果有大量重复,我们已经看到了具有1000万个reads的优秀RNA-seq实验。
8.差异表达分析工作流程
为了在进行差异表达分析时适当地对计数进行建模,已经开发了许多用于RNA-seq数据的差异表达分析的软件包。即使在不断开发新方法的过程中,通常也会建议使用一些工具作为最佳实践,例如DESeq2和EdgeR。这两种工具都使用负二项模型,采用类似的方法,通常产生类似的结果。它们非常严格,并且在灵敏度和特异性之间取得了良好的平衡(减少误报和假阴性)。
Limma-Voom是另一组常用于DE分析的工具,但这种方法对于小样本量可能不太敏感。当每组的生物学重复数量变大(> 20)时,推荐使用该方法。
许多描述这些方法之间比较的研究表明,尽管存在一些一致性,但工具之间也存在很大差异。此外,没有一种方法在所有条件下都能达到最佳效果(Soneson和Dleorenzi,2013)。

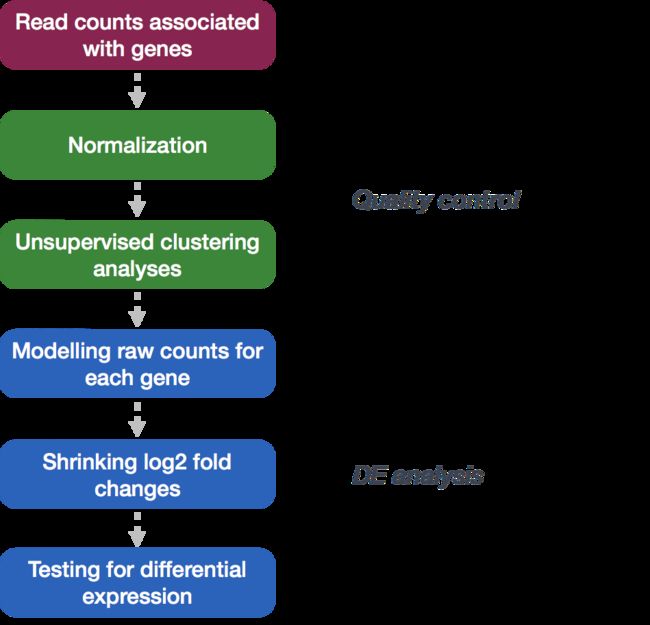
本课程将使用DESeq2进行DE分析,使用DESeq2的分析步骤将在下面的流程图中以绿色显示。DESeq2首先将计数数据标准化以解释样品之间文库大小和RNA组成的差异。然后用标准化计数在基因和样本水平上制作QC的一些图。最后一步是使用DESeq2包中的相应函数来执行差异表达分析。
接下来的课程中会深入介绍这些步骤,但有关DESeq2的更多详细信息和有用的建议可以在DESeq2的vignette中(http://bioconductor.org/packages/devel/bioc/vignettes/DESeq2/inst/doc/DESeq2.html)找到。如果完成此工作流程出现问题,可参考RStudio中的vignette:
vignette("DESeq2")
通过这个命令很方便地提供了触手可及的丰富信息!课程中如有需要可通过上述命令调用它。