原始数据查看:

grep '^@' ctr.R1.fq | sort |uniq|wc -l
27787625 也就是乘以四后的行数,即fq的name不会重
比对:
index=/media/pc/disk1/sun/refdata/ensembl_GRCm38/03_bowtie2_index/GRCm38
bowtie2 -p 20 -x $index -1 ctr.R1.fq -2 ctr.R2.fq -S ctr.sam &&
bowtie2 -p 20 -x $index -1 ko.R1.fq -2 ko.R2.fq -S ko.sam
比对结果:
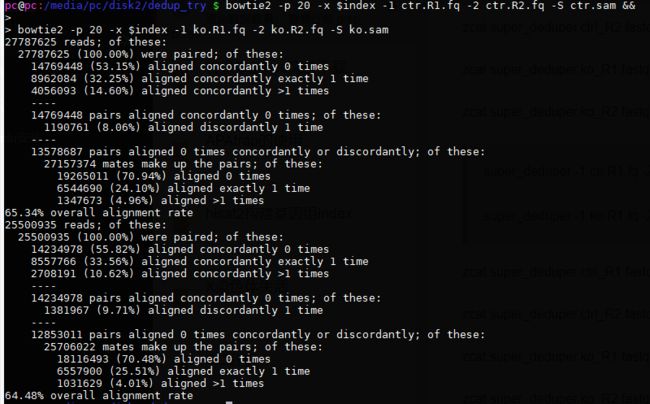
查看sam行数:

igv:
samtools sort --threads 10 -m 2G -o ctr.bam ctr.sam &&
samtools sort --threads 10 -m 2G -o ko.bam ko.sam
samtools index ctr.bam &&
samtools index ko.bam
提取mapped:
samtools view -h -@ 10 -F 4 ctr.sam > ctr.mapped.sam &&
samtools view -h -@ 10 -F 4 ko.sam > ko.mapped.sam
wc -l 查看mapped的sam行数:
对mapped的进行igv:
samtools sort --threads 10 -m 3G -o ctr.mapped.bam ctr.mapped.sam &&
samtools sort --threads 10 -m 3G -o ko.mapped.bam ko.mapped.sam &&
samtools index ctr.mappedbam &&
samtools index ko.mapped.bam
#################################
trim:
trimmomatic PE -phred33 -threads 20 ctr.R1.fq ctr.R2.fq ctr.R1.paired.fq ctr.R1.unpaired.fq ctr.R2.paired.fq ctr.R2.unpaired.fq ILLUMINACLIP:/home/pc/miniconda3/pkgs/trimmomatic-0.38-1/share/trimmomatic-0.38-1/adapters/TruSeq3-PE.fa:2:30:10:8:true LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 AVGQUAL:20 MINLEN:36
trimmomatic PE -phred33 -threads 20 ko.R1.fq ko.R2.fq ko.R1.paired.fq ko.R1.unpaired.fq ko.R2.paired.fq ko.R2.unpaired.fq ILLUMINACLIP:/home/pc/miniconda3/pkgs/trimmomatic-0.38-1/share/trimmomatic-0.38-1/adapters/TruSeq3-PE.fa:2:30:10:8:true LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 AVGQUAL:20 MINLEN:36
比对:
bowtie2 -p 20 -x $index -1 ctr.R1.paired.fq -2 ctr.R2.paired.fq -S ctr.paired.sam &&
bowtie2 -p 20 -x $index -1 ko.R1.paired.fq -2 ko.R2.paired.fq -S ko.paired.sam
经过trim后比对情况并未改善:

1.fatsquniq:
网站:https://sourceforge.net/projects/fastuniq/
用法:
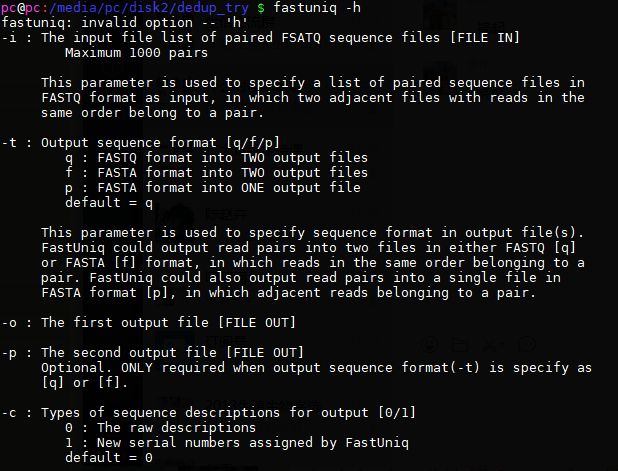
去重命令:
fastuniq -i list -o fastuniq.ctr.R1.fq -p fastuniq.ctr.R2.fq
fastuniq -i list2 -o fastuniq.ko.R1.fq -p fastuniq.ko.R2.fq
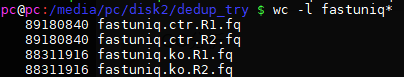
grep '^@' fastuniq.ctr.R1.fq | sort | uniq | wc -l
22295210 乘以四后也是行数;
去重率:
ctrl:80.23%
ko:86.58%
比对:
bowtie2 -p 20 -x $index -1 fastuniq.ctr.R1.fq -2 fastuniq.ctr.R2.fq -S fastuniq.ctr.sam &&
bowtie2 -p 20 -x $index -1 fastuniq.ko.R1.fq -2 fastuniq.ko.R2.fq -S fastuniq.ko.sam
比对结果:

查看sam:
44590443 fastuniq.ctr.sam
44155981 fastuniq.ko.sam
igv:
samtools sort --threads 10 -m 2G -o fastuniq.ctr.bam fastuniq.ctr.sam &&
samtools sort --threads 10 -m 2G -o fastuniq.ko.bam fastuniq.ko.sam &&
samtools index fastuniq.ctr.bam &&
samtools index fastuniq.ko.bam
提取mapped:
samtools view -h -@ 10 -F 4 fastuniq.ctr.sam > fastuniq.ctr.mapped.sam &&
samtools view -h -@ 10 -F 4 fastuniq.ko.sam > fastuniq.ko.mapped.sam
查看mapped行数:
提取mapped后不能用igv。原因是缺少header:
解决:提取mapped时加-h参数:ref:[E::sam_parse1] missing SAM header
2.seqkit:
网站:https://bioinf.shenwei.me/seqkit/
用法:
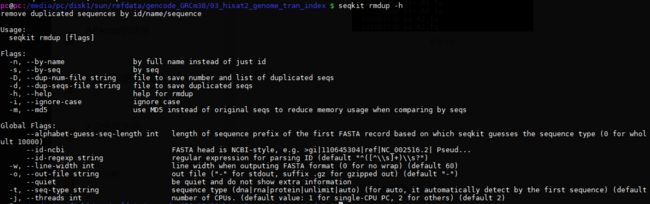
命令:
seqkit rmdup -s ctr.R1.fq -o seqkit.ctr.R1.fq.gz -d seqkit.ctr.R1.duplicated.fq.gz -D seqkit.ctr.R1.duplicated.txt -j 5
seqkit rmdup -s ctr.R2.fq -o seqkit.ctr.R2.fq.gz -d seqkit.ctr.R2.duplicated.fq.gz -D seqkit.ctr.R2.duplicated.txt -j 5
seqkit rmdup -s ko.R1.fq -o seqkit.ko.R1.fq.gz -d seqkit.ko.R1.duplicated.fq.gz -D seqkit.ko.R1.duplicated.txt -j 5
seqkit rmdup -s ko.R2.fq -o seqkit.ko.R2.fq.gz -d seqkit.ko.R2.duplicated.fq.gz -D seqkit.ko.R2.duplicated.txt -j 5
查看行数:
zcat seqkit.ctr.R1.fq.gz | wc -l -> 57594872 51.82% [去重]
zcat seqkit.ctr.R2.fq.gz | wc -l -> 63321348 56.97% [去重]
zcat seqkit.ko.R1.fq.gz | wc -l -> 60717652 54.63% [去重]
zcat seqkit.ko.R2.fq.gz | wc -l -> 67585076 66.26% [去重]
比对:
bowtie2 -p 20 -x $index -1 seqkit.ctr.R1.fq.gz -2 seqkit.ctr.R2.fq.gz -S seqkit.ctr.sam &&
bowtie2 -p 20 -x $index -1 seqkit.ko.R1.fq -2 seqkit.ko.R2.fq -S seqkit.ko.sam
报错了:双端不对齐
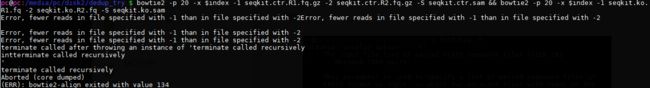
结论:
seqkit去重率约50-70%,但是使用后双端不齐,无法比对。但是可以保留去重掉的fastq。
3.super_deduper:
官网:https://github.com/dstreett/Super-Deduper
新地址:https://github.com/ibest/HTStream
用法:
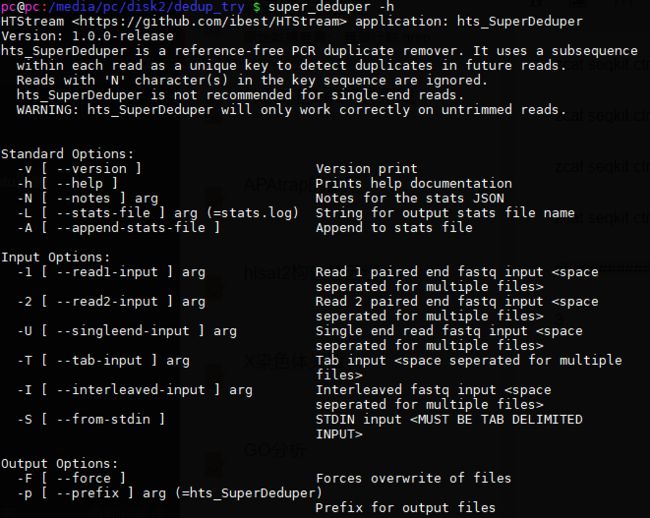
命令:
super_deduper -1 ctr.R1.fq -2 ctr.R1.fq -g -p super_deduper.ctrl
super_deduper -1 ko.R1.fq -2 ko.R1.fq -g -p super_deduper.ko
查看行数:
zcat super_deduper.ctrl_R1.fastq.gz |wc -l -> 3915060 3.52% [去重]
zcat super_deduper.ctrl_R2.fastq.gz |wc -l -> 3915060 3.52% [去重]
zcat super_deduper.ko_R1.fastq.gz |wc -l -> 3971360 3.89% [去重]
zcat super_deduper.ko_R2.fastq.gz |wc -l -> 3971360 3.89% [去重]
修改参数:
super_deduper -1 ctr.R1.fq -2 ctr.R1.fq -g -p super_deduper.ctrl -q 30
super_deduper -1 ko.R1.fq -2 ko.R1.fq -g -p super_deduper.ko -q 20
查看again:
zcat super_deduper.ctrl_R1.fastq.gz | wc -l -> 3915060 3.52% [去重]
zcat super_deduper.ctrl_R2.fastq.gz | wc -l -> 3915060
zcat super_deduper.ko_R1.fastq.gz | wc -l -> 3971360 3.89% [去重]
zcat super_deduper.ko_R2.fastq.gz | wc -l -> 3971360 改了参数后竟然没变
比对:
bowtie2 -p 20 -x $index -1 super_deduper.ctrl_R1.fastq.gz -2 super_deduper.ctrl_R2.fastq.gz -S super_deduper.ctrl.sam &&
bowtie2 -p 20 -x $index -1 super_deduper.ko_R1.fastq.gz -2 super_deduper.ko_R2.fastq.gz -S super_deduper.ko.sam
比对结果:
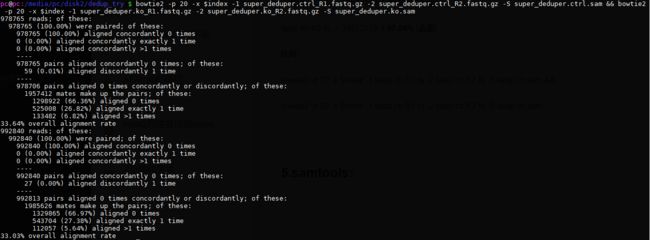
查看sam行数:
igv:
samtools sort --threads 10 -m 2G -o super_deduper.ctrl.bam super_deduper.ctrl.sam && samtools sort --threads 10 -m 2G -o super_deduper.ko.bam super_deduper.ko.sam
samtools index super_deduper.ctrl.bam &&
samtools index super_deduper.ko.bam
提取mapped:
samtools view -h -@ 10 -F 4 super_deduper.ctrl.sam > super_deduper.ctrl.mapped.sam &&
samtools view -h -@ 10 -F 4 super_deduper.ko.sam > super_deduper.ko.mapped.sam
查看mapped的行数
结论:
super_deduper率约3-4%,去重去的太多。原理是什么呢?
4.fastp:
官网:https://github.com/OpenGene/fastp
用法:

命令:
fastp -i ctr.R1.fq -I ctr.R2.fq -o fastp.ctr.R1.fq -O fastp.ctr.R2.fq
fastp -i ko.R1.fq -I ko.R2.fq -o fastp.ko.R1.fq -O fastp.ko.R2.fq
查看:

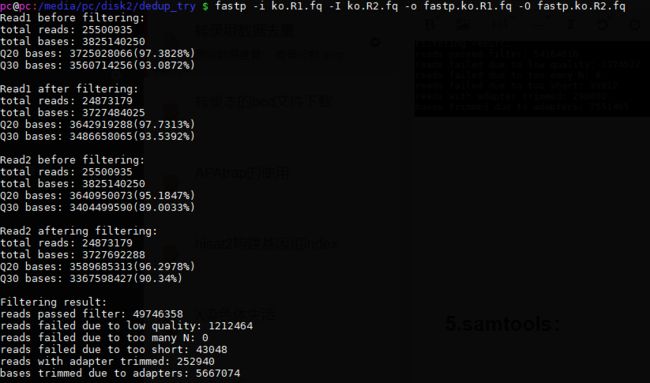
查看:
fastp.ctr.R1.fq -> 27082408*4 97.46% [去重]
fastp.ctr.R2.fq -> 27082408*4 97.46% [去重]
fastp.ko.R1.fq -> 24873179*4 97.54% [去重]
fastp.ko.R2.fq -> 24873179*4 97.54% [去重]
比对:
bowtie2 -p 20 -x $index -1 fastp.ctr.R1.fq -2 fastp.ctr.R2.fq -S fastp.ctr.sam &&
bowtie2 -p 20 -x $index -1 fastp.ko.R1.fq -2 fastp.ko.R2.fq -S fastp.ko.sam
比对情况:

提取mapped:
samtools view -h -@ 10 -F 4 fastp.ctr.sam > fastp.ctr.mapped.sam &&
samtools view -h -@ 10 -F 4 fastp.ko.sam > fastp.ko.mapped.sam
查看行:
35,942,791 fastp.ctr.mapped.sam
32,536,623 fastp.ko.mapped.sam
igv:
samtools sort --threads 10 -m 2G -o fastp.ctr.mapped.bam fastp.ctr.mapped.sam&& samtools sort --threads 10 -m 2G -o fastp.ko.mapped.bamfastp.ko.mapped.sam &&
samtools index fastp.ctr.mapped.bam &&
samtools index fastp.ko.mapped.bam
5.samtools:
官网:http://www.htslib.org/
samtools sort --threads 10 -n ctr.bam -o ctr.sort.bam &&
samtools fixmate --threads 10 -m ctr.sort.bam ctr.fixmate.bam &&
samtools sort --threads 10 ctr.fixmate.bam -o ctr.positionsort.bam &&
samtools markdup --threads 10 -r ctr.positionsort.bam ctr.markdup.bam
samtools sort --threads 10 -n ko.bam -o ko.sort.bam &&
samtools fixmate --threads 10 -m ko.sort.bam ko.fixmate.bam &&
samtools sort --threads 10 ko.fixmate.bam -o ko.positionsort.bam &&
samtools markdup --threads 10 -r ko.positionsort.bam ko.markdup.bam
查看行:
samtools view ctr.markdup.bam | wc -l 39,334,011
samtools view ko.markdup.bam | wc -l 39,170,695
提取mapped:
samtools view -h -@ 10 -F 4 ctr.markdup.bam > ctr.markdup.mapped.sam &&
samtools view -h -@ 10 -F 4 ko.markdup.bam > ko.markdup.mapped.sam
查看mapped的行数:
wc -l ctr.markdup.mapped.sam -> 20,069,023
wc -l ko.markdup.mapped.sam -> 21,054,225
igv:
samtools sort --threads 10 -m 2G -o ctr.markdup.mapped.bam ctr.markdup.mapped.sam && samtools sort --threads 10 -m 2G -o ko.markdup.mapped.bam ko.markdup.mapped.sam &&
samtools index ctr.markdup.mapped.bam &&
samtools index ko.markdup.mapped.bam
6.picard:
官网:https://broadinstitute.github.io/picard/
用法:

直接删除冗余:
java -jar /home/pc/biosoft/picard.jar MarkDuplicates REMOVE_DUPLICATES=true I=ctr.sort.bam O=picard.ctr.sorted.markdup.bam M=picard.ctr.markdup.txt
****************************************************
java -jar /home/pc/biosoft/picard.jar MarkDuplicates REMOVE_DUPLICATES=true I=ko.sort.bam O=picard.ko.sorted.markdup.bam M=picard.ko.markdup.txt
查看行:
samtools view picard.ctr.sorted.markdup.bam | wc -l -> 39,302,611
samtools view picard.ko.sorted.markdup.bam | wc -l -> 39,151,069
igv:
samtools index picard.ctr.sorted.markdup.bam
samtools index picard.ko.sorted.markdup.bam
提取mapped:
samtools view -h -@ 10 -F 4 picard.ctr.sorted.markdup.bam > picard.ctr.sorted.markdup.mapped.sam &&
samtools view -h -@ 10 -F 4 picard.ko.sorted.markdup.bam > picard.ko.sorted.markdup.mapped.sam
查看行数:
20,037,624 picard.ctr.sorted.markdup.mapped.sam
21,034,600 picard.ko.sorted.markdup.mapped.sam
igv:
samtools sort --threads 10 -m 2G -o picard.ctr.sorted.markdup.mapped.bam picard.ctr.sorted.markdup.mapped.sam &&
samtools sort --threads 10 -m 2G -o picard.ko.sorted.markdup.mapped.bam picard.ko.sorted.markdup.mapped.sam &&
samtools index picard.ctr.sorted.markdup.mapped.bam &&
samtools index picard.ko.sorted.markdup.mapped.bam
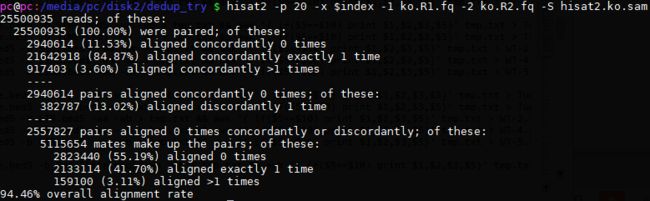
# 比对效率低?
用hisat2比对看竟然比对效率提高这么多