#上期我们比较了TCGA肝癌组织 vs 正常组织的差异表达分析.但由于它的正常样本也来自于肝癌病人可能导致样本聚类混乱.本期开始使用GTEx的正常Liver样本数据来代替TCGA的正常样本数据进行差异表达分析.来挖掘与TCGA肝癌病人的正常样本的差异.如果与之前的分析有较大差异,并且更符合实验验证结果,那说明TCGA正常样本的数据可能是不适合作为差异表达分析的对照组的.
GTEx的表达谱数据来源于175名死者身上采集的1641个尸检样本,这些样本来自身体43个不同的部位.包含5万4千个基因的测序信息.
我们首先下载GTEx的样本注释文件和表达谱文件,链接如下: https://www.gtexportal.org/home/datasets .上面的是注释文件,下面的是表达谱.
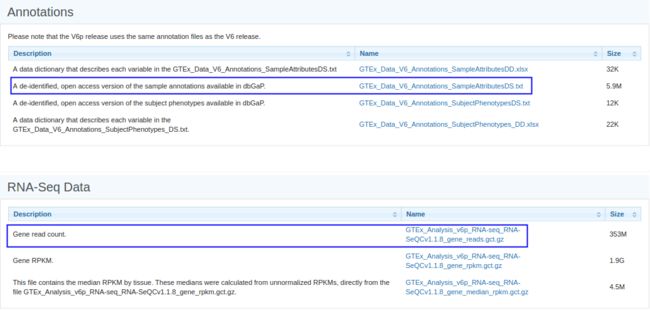
然后进行提取正常Liver组织的表达谱:
# 获取所有liver样本的ID:
awk 'BEGIN{OFS="\t";FS="\t"}{
if(NR==1){
for(i=1;i1 && $target_id=="Liver")print $0;
}' ./GTEx_Data_V6_Annotations_SampleAttributesDS.txt >./GTEx_Data_V6_Anno_liver.tsv
提取出143个Liver的样本,第一列是ID,我们需要凭ID从所有组织样本表达谱文件中提取Liver的.
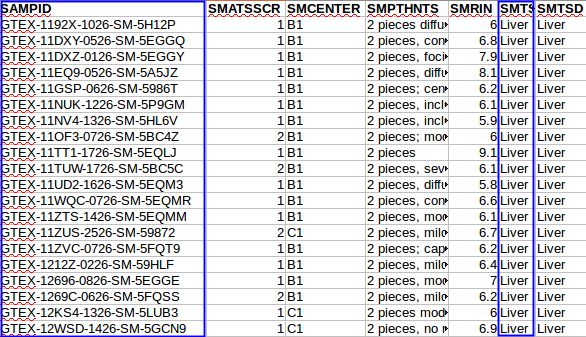
将表达谱文件解压:
# 解压:
gunzip -d GTEx_Analysis_v6p_RNA-seq_RNA-SeQCv1.1.8_gene_reads.gct.gz
解压后发现还是很大,有1.3个G,那我们看看前1000行是啥样子:
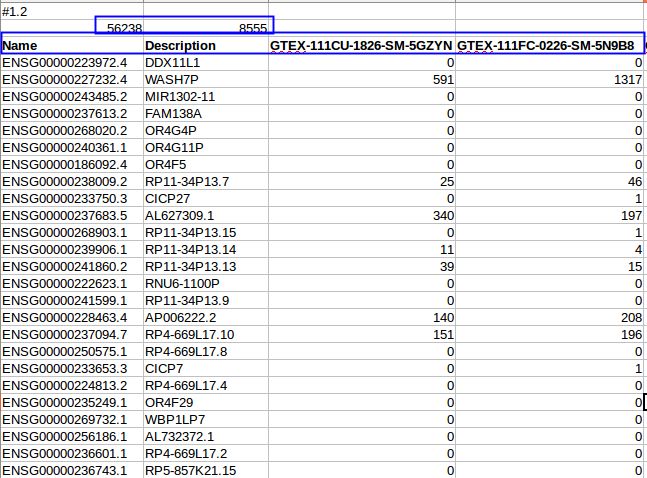
前两行是注释,第二行是说有56238个"gene"和8555个样本,从第三行开始是样本ID所对应的"gene"的表达量.这56238个"gene"里囊括所有的蛋白编码基因,lncRNA,pseudogene,miRNA等所有"gene".我们先根据列名提取Liver所对应ID的样本表达谱:
awk 'BEGIN{OFS="\t";FS="\t"}{
#处理第一个文件:
if(NR==FNR && NR>1){a[$1];} #保存第一列ID到数组a
#处理第二个文件:
if(NR>FNR && FNR==3){
printf "%s\t%s",$1,$2; #输出第一列"Name"和"Description"列名
for(i=3;i<=NF;i++){
if($i in a){
b[i]=$i; #保存ID到下标为列数的数组b
printf "\t%s",$i; #输出ID
}
}
printf "\n";
#由于awk自动输出无序数组,所以对数组排序,保存排序后b的下标到bs
bl=asorti(b,bs);
#排序输出:for(i=1;i<=bl;i++)print "i:"i"\tbs:"bs[i]"\tbsi"b[bs[i]];
}
if(NR>FNR && FNR>4){
printf "%s\t%s",$1,$2; #输出第一列gene_ID和gene_name列名
for(i=1;i<=bl;i++){
printf "\t%s",$bs[i]; #bs保存了排序后的列号;
}
printf "\n";
}
}' ./GTEx_Data_V6_Anno_liver.tsv ./GTEx_Analysis_v6p_RNA-seq_RNA-SeQCv1.1.8_gene_reads.gct >./GTEx_Analysis_v6p_RNA_Liver.tsv
结果文件GTEx_Analysis_v6p_RNA_Liver.tsv部分内容如下:
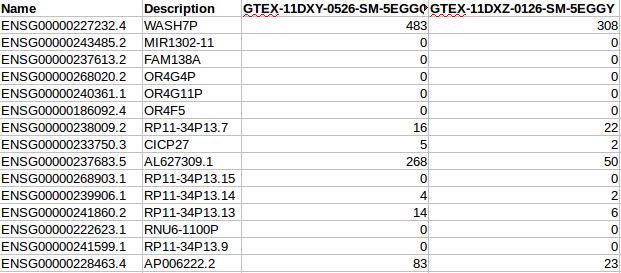
但是我们发现一个问题,我们提取出来的Liver的样本ID有143个,但我们从总表达谱文件中只提取到119个样本的数据,也就是说有24个样本通过上面的脚本没有提取出来,仔细检查了脚本是没有问题的.
wc -l ./GTEx_Data_V6_Anno_liver.tsv
# 结果为144,除去首航列名后是143.
awk 'BEGIN{FS="\t"}{if(NR==1)print NF}' ./GTEx_Analysis_v6p_RNA_Liver.tsv
# 结果为121,去除掉前两列非样本列后只有119列.
然后我直接去看之前提取的Liver的样本注释文件GTEx_Analysis_v6p_RNA_Liver.tsv, 发现后面有部分样本描述的信息缺失,并且数目也刚好是24个.这会不会是我们提取样本数目对不上的原因呢?
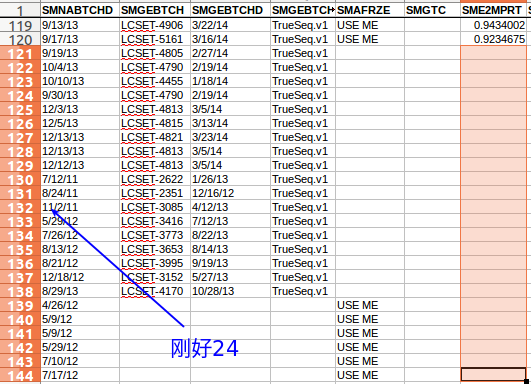
为了查看这24个样本缺失了什么属性值,我们从GTEx的下载页面下载关于列属性的描述文件:
我们发现24个样本缺失的都是如下属性值,reads的map率, 嵌合reads,基因外reads比例,空位数目等关于测序结果的描述.这24个样本缺少这些信息表明它们可能没有测序或者质量不佳,所以没有被收录在,这里不做深究,也就是说我们只能从143个Liver样本中获得119个样本的表达谱.

上面是使用shell的awk(我的最爱)从1.3个G的表达谱矩阵中提取想要的样本数据,其实对于这种矩阵类型的文件,R更方便逻辑更清晰,代码如下:
# 初始化环境:
rm(list=ls())
# 载入包:
library(data.table)
# 注释文件路径:
GTEx_liver_anno_path<-"./GTEx_Data_V6_Anno_liver.tsv"
# 读入注释文件:
GTEx_liver_anno<-fread(GTEx_liver_anno_path,sep = "\t",header = T)
# 获得要提取的样本ID:
liver_ID<-GTEx_liver_anno[,SAMPID]
# 再添加两个列名:
liver_ID<-append(c("Name","Description"),liver_ID)
# 1.3G的表达谱文件路径:
GTExFile_path<-"./GTEx_Analysis_v6p_RNA-seq_RNA-SeQCv1.1.8_gene_reads.gct"
#根据要提取的样本ID读入文件,skip前两行,select要读取的列,header是要的,分隔符是Tab.
GTEx_reads<-fread(GTExFile_path,sep="\t",header =T,skip = 2,select = liver_ID)
# 输出文件:
fwrite(GTEx_reads,"./GTEx_Analysis_v6p_RNA_Liver2.tsv",sep = "\t")
对于1.3G这种还比较大的矩阵格式比较工整的文件,从逻辑来看,R更方便过程更容易理解,但AWK更快一点.本期先完成文件提取整合,下期开始进行miRNA ID转换及分析.
更多原创精彩内容敬请关注生信杂谈:
