但如果没条件的话,可以尝试利用现有的数据(比如:千人基因组项目,GIAB http://jimb.stanford.edu/giab/等)复现它们的成果,甚至只是构建一个分析流程也行,这样子学起来才会比较高效,同时也有利于夯实所学的知识。
找例子:数据库:ATGG
流程图:
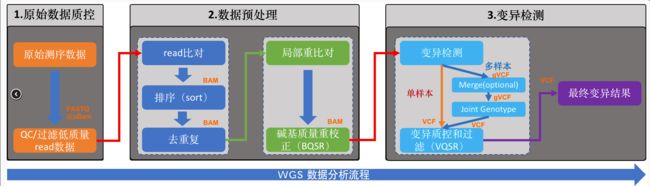
常用软件:bwa,samtools,picard,GATK,bedtools,bcftools,vcftools,FastQC,MultiQC,VEP
例子:http://rosalind.info/problems/locations/ 包括:RNA,DNA,蛋白
GWAS
全基因组测序分布-China

软件:比对软件-BWA;
数据转换-samtools
- Picard
基因数据变异检测工具-GATK
[if !supportLists]1. [endif]质控:
软件FASTQC质控,
1. [endif]read各个位置的碱基质量值分布
2. [endif]碱基的总体质量值分布
3. [endif]read各个位置上碱基分布比例,目的是为了分析碱基的分离程度
4. [endif]GC含量分布
5. [endif]read各位置的N含量
6. [endif]read是否还包含测序的接头序列
7. [endif]read重复率,这个是实验的扩增过程所引入的
2. 比对
BWA比对

通常把同一个样本的所有比对结果变成一个:因为有的测序深度深或者需要不同的lane来测序; $ samtools merge [ ...]
gatk \
-T RealignerTargetCreator \ ##目的是定位出所有需要进行序列重比对的目标区域
-R /path/to/human.fasta\
-Isample_name.sorted.markdup.bam \
-known/path/to/gatk/bundle/1000G_phase1.indels.b37.vcf \
-known/path/to/gatk/bundle/Mills_and_1000G_gold_standard.indels.b37.vcf \
-osample_name.IndelRealigner.intervals
Gatk \
-T IndelRealigner \ ## 对所有在第一步中找到的目标区域运用算法进行序列重比对,最后得到捋顺了的新结果。
-R /path/to/human.fasta \
-I sample_name.sorted.markdup.bam \
-known /path/to/gatk/bundle/1000G_phase1.indels.b37.vcf\
-known /path/to/gatk/bundle/Mills_and_1000G_gold_standard.indels.b37.vcf \
-o sample_name.sorted.markdup.realign.bam \
--targetIntervals sample_name.IndelRealigner.intervals
注意:1000G_phase1.indels.b37.vcf和Mills_and_1000G_gold_standard.indels.b37.vcf。这两个文件来自于千人基因组和Mills项目,里面记录了那些项目中检测到的人群Indel区域。
重新校正碱基质量值(BQSR)Base Quality Score Recalibration
主要是通过机器学习的方法构建测序碱基的错误率模型,然后对这些碱基的质量值进行相应的调整。
流程:
gatk \
-T BaseRecalibrator \
-R /path/to/human.fasta \
-I sample_name.sorted.markdup.realign.bam \
--knownSites /path/to/gatk/bundle/1000G_phase1.indels.b37.vcf \
--knownSites /path/to/gatk/bundle/Mills_and_1000G_gold_standard.indels.b37.vcf \
--knownSites /path/to/gatk/bundle/dbsnp_138.b37.vcf \
-o sample_name.recal_data.table
###BaseRecalibrator,这里计算出了所有需要进行重校正的read和特征值,然后把这些信息输出为一份校准表文件(sample_name.recal_data.table)
gatk
-T PrintReads \
-R /path/to/human.fasta \
-I sample_name.sorted.markdup.realign.bam \
--BQSR sample_name.recal_data.table \
-o sample_name.sorted.markdup.realign.BQSR.bam
##PrintReads,这一步利用第一步得到的校准表文件(sample_name.recal_data.table)重新调整原来BAM文件中的碱基质量值,并使用这个新的质量值重新输出一份新的BAM文件。
变异检测 SNP、Indel,CNV和SV等
方法:使用GATK
HaplotypeCaller模块对样本中的变异进行检测,它也是目前最适合用于对二倍体基因组进行变异(SNP+Indel)检测的算法。
一般来说,在实际的WGS流程中对HaplotypeCaller的应用有两种做法,差别只在于要不要在中间生成一个gVCF:
(1)直接进行HaplotypeCaller,这适合于单样本,或者那种固定样本数量的情况,也就是执行一次HaplotypeCaller之后就老死不相往来了。否则你会碰到仅仅只是增加一个样本就得重新运行这个HaplotypeCaller的坑爹情况(即,N+1难题),而这个时候算法需要重新去读取所有人的BAM文件,这将会是一个很费时间的痛苦过程;
gatk \
-T HaplotypeCaller \
-R /path/to/human.fasta \
-I sample_name.sorted.markdup.realign.BQSR.bam \
-D /path/to/gatk/bundle/dbsnp_138.b37.vcf \
-stand_call_conf 50 \
-A QualByDepth \
-A RMSMappingQuality \
-A MappingQualityRankSumTest \
-A ReadPosRankSumTest \
-A FisherStrand \
-A StrandOddsRatio \
-A Coverage \
-o sample_name.HC.vcf
这里我特别提一下-D参数输入的dbSNP同样可以再GATK bundle目录中找到,这份文件汇集的是目前几乎所有的公开人群变异数据集。另外,由于我们的例子只有一个样本因此只输入一个BAM文件就可以了,如果有多个样本那么可以继续用-I参数输入:
(2)每个样本先各自生成gVCF,然后再进行群体joint-genotype。这其实就是GATK团队为了解决(1)中的N+1难题而设计出来的模式。gVCF全称是genome VCF,是每个样本用于变异检测的中间文件,格式类似于VCF,它把joint-genotype过程中所需的所有信息都记录在这里面,文件无论是大小还是数据量都远远小于原来的BAM文件。这样一旦新增加样本也不需要再重新去读取所有人的BAM文件了,只需为新样本生成一份gVCF,然后重新执行这个joint-genotype就行了。
gatk \
-T HaplotypeCaller \
-R reference.fasta \
-I sample1.bam [-I sample2.bam ...] \
...一样了
然而,基因组上各个不同的染色体之间其实是可以理解为相互独立的(结构性变异除外),也就是说,为了提高效率我们可以按照染色体一条条来独立执行这个步骤,最后再把结果合并起来就好了,这样的话就能够节省很多的时间。下面我给出一个按照染色体区分的例子:
gatk \
-T HaplotypeCaller \
-R /path/to/human.fasta \
-I sample_name.sorted.markdup.realign.BQSR.bam\
-D /path/to/gatk/bundle/dbsnp_138.b37.vcf \
-L 1 \
-stand_call_conf 50 \
-A QualByDepth \
-A RMSMappingQuality \
-A MappingQualityRankSumTest \
-A ReadPosRankSumTest \
-A FisherStrand \
-A StrandOddsRatio \
-A Coverage \
-o sample_name.HC.1.vcf
注意到了吗?其它参数都没任何改变,就只增加了一个 -L 参数,通过这个参数我们可以指定特定的染色体(或者基因组区域)!我们这里指定的是 1 号染色体,有些地方会写成chr1,具体看human.fasta中如何命名,与其保持一致即可。其他染色体的做法也是如此,就不再举例了。最后合并:
gatk \
-T CombineVariants \
-R /path/to/human.fasta \
--genotypemergeoption UNSORTED \
--variant sample_name.HC.1.vcf \
--variant sample_name.HC.2.vcf \
...
--variant sample_name.HC.MT.vcf \
-o sample_name.HC.vcf
第二种,先产生gVCF,最后再joint-genotype的做法:
java -jar /path/to/GenomeAnalysisTK.jar \
-THaplotypeCaller \
-R/path/to/human.fasta \
-Isample_name.sorted.markdup.realign.BQSR.bam \
--emitRefConfidence GVCF \
-osample_name.g.vcf
#调用GenotypeGVCFs完成变异calling
java -jar /path/to/GenomeAnalysisTK.jar \
-TGenotypeGVCFs \
-R/path/to/human.fasta \
--variant sample_name.g.vcf \
-osample_name.HC.vcf
其实,就是加了–emitRefConfidence GVCF的参数。而且,假如嫌慢,同样可以按照染色体或者区域去产生一个样本的gVCF,然后在GenotypeGVCFs中把它们全部作为输入文件完成变异calling。也许你会担心同个样本被分成多份gVCF之后,是否会被当作不同的多个样本?回答是不会!因为生成gVCF文件的过程中,GATK会根据@RG信息中的SM(也就是sample name)来判断这些gVCF是否来自同一个样本,如果名字相同,那么就会被认为是同一个样本,不会产生多样本问题。
变异检测质控和过滤(VQSR)
这是我们这个流程中最后的一步了。在获得了原始的变异检测结果之后,我们还需要做的就是质控和过滤。这一步或多或少都有着一些个性化的要求,我暂时就不做太多解释吧(一旦解释恐怕同样是一篇万字长文)。只用一句话来概括,VQSR是通过构建GMM模型对好和坏的变异进行区分,从而实现对变异的质控,具体的原理暂时不展开了。
下面就直接给出例子吧:
## SNP Recalibrator
java -jar /path/to/GenomeAnalysisTK.jar \
-TVariantRecalibrator \
-Rreference.fasta \
-input sample_name.HC.vcf \
-resource:hapmap,known=false,training=true,truth=true,prior=15.0/path/to/gatk/bundle/hapmap_3.3.b37.vcf \
-resource:omini,known=false,training=true,truth=false,prior=12.0/path/to/gatk/bundle/1000G_omni2.5.b37.vcf \
-resource:1000G,known=false,training=true,truth=false,prior=10.0/path/to/gatk/bundle/1000G_phase1.snps.high_confidence.b37.vcf \
-resource:dbsnp,known=true,training=false,truth=false,prior=6.0/path/to/gatk/bundle/dbsnp_138.b37.vcf \
-an QD -an MQ -an MQRankSum -an ReadPosRankSum -an FS -an SOR -an DP \
-mode SNP \
-recalFile sample_name.HC.snps.recal \
-tranchesFile sample_name.HC.snps.tranches \
-rscriptFile sample_name.HC.snps.plots.R
java -jar /path/to/GenomeAnalysisTK.jar -TApplyRecalibration \
-R human_g1k_v37.fasta \
-input sample_name.HC.vcf \
--ts_filter_level 99.5 \
-tranchesFile sample_name.HC.snps.tranches \
-recalFile sample_name.HC.snps.recal \
-mode SNP \
-osample_name.HC.snps.VQSR.vcf
## Indel Recalibrator
java -jar /path/to/GenomeAnalysisTK.jar -TVariantRecalibrator \
-R human_g1k_v37.fasta \
-input sample_name.HC.snps.VQSR.vcf \
-resource:mills,known=true,training=true,truth=true,prior=12.0/path/to/gatk/bundle/Mills_and_1000G_gold_standard.indels.b37.vcf \
-an QD -an DP -an FS -an SOR -an ReadPosRankSum -an MQRankSum \
-mode INDEL \
-recalFile sample_name.HC.snps.indels.recal \
-tranchesFile sample_name.HC.snps.indels.tranches \
-rscriptFile sample_name.HC.snps.indels.plots.R
java -jar /path/to/GenomeAnalysisTK.jar -TApplyRecalibration \
-Rhuman_g1k_v37.fasta\
-input sample_name.HC.snps.VQSR.vcf \
--ts_filter_level 99.0 \
-tranchesFile sample_name.HC.snps.indels.tranches \
-recalFile sample_name.HC.snps.indels.recal \
-mode INDEL \
-osample_name.HC.snps.indels.VQSR.vcf
最后,sample_name.HC.snps.indels.VQSR.vcf便是我们最终的变异检测结果。对于人类而言,一般来说,每个人最后检测到的变异数据大概在400万左右(包括SNP和Indel)。
http://www.huangshujia.me/2017/09/19/2017-09-19-Begining-WGS-Data-Analysis-The-pipeline.html
例子:http://starsyi.github.io/2016/05/24/BWA-%E5%91%BD%E4%BB%A4%E8%AF%A6%E8%A7%A3/