1 介绍
基因组重测序是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。
基于全基因组重测序技术,人们期望快速进行资源普查筛选,寻找到大量遗传变异,实现遗传进化分析及重要性状候选基因的预测。随着测序成本降低和拥有参考基因组序列物种增多,全基因组重测序成为动植物育种和群体进化研究迅速有效的方法。
• SNP 强调群体在这个位点有多样性。
• Genotype 强调个体在这个位点是哪种基因型。SNV属于genotype的范畴,表示这个genotype 是个突变,是第一无二的。
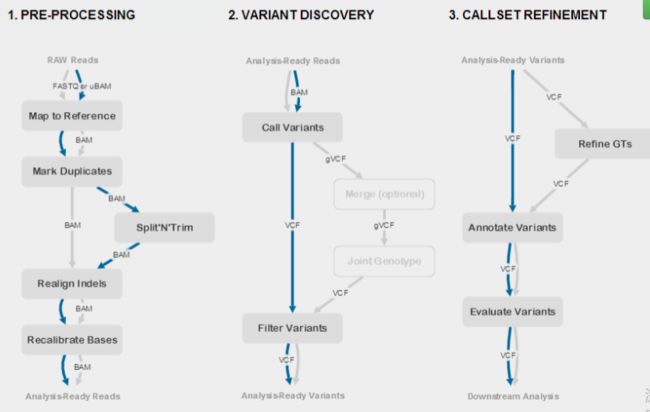

2 基因组SNP calling分析流程
• 首先利用bwa,samtools,picard对参考基因组ref.fa建立索引,随后使用基因组比对软件bwa将fastq文件比对回参考基因组,生成sam文件(这里使用例子数据由于小于10M,所以使用bwa的-is参数,当参考基因组很大的时候,使用bwtsw参数)
• 使用picard对sam文件排序后生成bam文件(这里如果使用samtools的话会删除重复位点,而picard只会标记重复并不会删除)。之后使用samtools对bam文件建立索引,利用已经注释过的indel.vcf文件对indel周围进行realignment,这个操作有两个步骤,第一步生成需要进行realignment的位置的信息,第二步 才是对这些位置进行realignment,最后生成一个重排好的BAM。
• 接下来对mapping得到的bam文件进行矫正,消除偏态,去除假阳性,这里主要针对测序质量不是特别好的数据,可以根据fastqc结果对测序结果进行解读,如果比较好的话可以跳过。
• 使用HaplotypeCaller发现变异并生成gvcf文件。随后使用CombineGVCFs将不同文件整合,GenotypeGVCFs函数输出vcf文件。
• 下一步使用VariantRecalibrator校准,并根据可视化结果确定最佳参数,确定参数之后再运行一次VariantRecalibrator得出结果
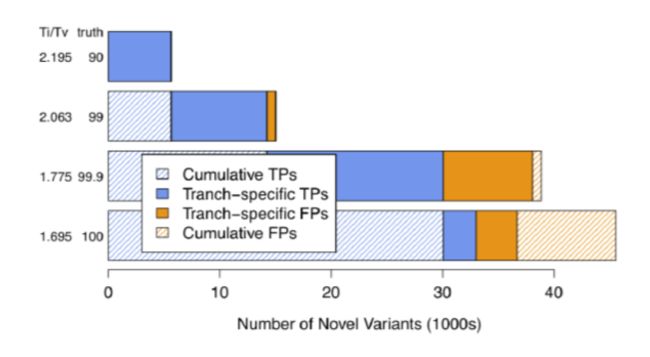
• 如何确定参数:(1) an注释信息选择:从图中可以看出,这个模拟结果可以很好的将真实的变异位点和假阳性变异位点分开,形成了明显的界限, 也就是说,如果一个变异位点的这两个注释值,只要有一个落在了界限之外,就会被过滤掉。最主要的是要 看右边两个图片,只要能很好的区分开novel和known以及filtered和retained就可以。其实在如何选择注释值 存在一定得主观性,因此,在做VariantRecalibrator时可以做两次,第一次尽可能的多的选择这些注释值, 第一遍跑完之后,选择几个区分好的,再做一次VariantRecalibrator,然后再做ApplyRecalibration。 (2) tranche值的设定:如果这个值设定的比较高的话,那么最后留下来的变异位点就会多,但同时假阳性的位点也会相应增加; run两遍VariantRecalibrator,第一遍的时候多写几个阈值,看哪个阈值结果好,选择一个最好的阈值,再run一遍 VariantRecalibrator。 区分标准: 1. 看结果中已知变异位点与新发现变异位点之间的比例,这个比例不要太大,大多数新发现的变异都是假阳性,如果太多的 话,可能假阳性的比例就比较大; 2. 看保留的变异数目,这个就要根据具体的需求进行选择了。 – 3. 看TI/TV值,对于人类全基因组,这个值应该在2.15左右,对于外显子组,这个值应该在3.2左右,不要太小或太大,越接近 这个数值越好,这个值如果太小,说明可能存在比较多的假阳性。 千人中所选择的tranche值是99,仅供参考。
• 最后使用annovar对vcf进行注释并可视化结果
3 代码展示
3.1建立索引
对于基因组
#首先建立参考基因组索引
##bwa
bwa index -a is ./ref/TAIR9_chr1.random.fasta
##samtools
samtools faidx ./ref/TAIR9_chr1.random.fasta
##picard
java -jar /software/picard-tools-1.119/CreateSequenceDictionary.jar \
REFERENCE=./ref/TAIR9_chr1.random.fasta\
OUTPUT=./ref/TAIR9_chr1.random.dict
对于转录组
###STAR建立索引
STAR -runThreadN 8 -runMode genomeGenerate \
-genomeDir ./star_index/ \
-genomeFastaFiles ./genome/chrX.fa \
-sjdbGTFfile ./genes/chrX.gtf
3.2 比对回参考基因组
基因组使用bwa
#bwa将测序文件比对回参考基因组
bwa mem -t 4 -M -R "@RG\tID:Sample12\tLB:Sample12\tPL:Illumina\tPU:Sample12\tSM:Sample12" \
./ref/TAIR9_chr1.random.fasta \
./reads/vars_20x/Sample12_1.fq\
./reads/vars_20x/Sample12_2.fq\
>./reads/vars_20x/Sample12.bwa.sam
##这里引号中表示对SAM 头文件添加标签
转录组使用STAR 2-pass,第一步比对
STAR -runThreadN 8 -genomeDir ./star_index/ \
-readFilesIn ./samples/ERR188044_chrX_1.fastq.gz ./samples/ERR188044_chrX_2.fastq.gz \
-readFilesCommand zcat \
-outFileNamePrefix ./star_1pass/ERR188044
根据比对结果重新建立索引
STAR -runThreadN 8 -runMode genomeGenerate \
-genomeDir ./star_index_2pass/ \
-genomeFastaFiles ./genome/chrX.fa \
-sjdbFileChrStartEnd ./star_1pass/ERR188044SJ.out.tab
进行第二次比对
STAR -runThreadN 8 -genomeDir ./star_index_2pass/ \
-readFilesIn ./samples/ERR188044_chrX_1.fastq.gz ./samples/ERR188044_chrX_2.fastq.gz \
-readFilesCommand zcat \
-outFileNamePrefix ./star_2pass/ERR188044
3.3 sort并转换为bam文件
基因组使用SortSam.jar
#picard排序并转换为bam文件
java -jar /software/picard-tools-1.119/SortSam.jar \
VALIDATION_STRINGENCY=SILENT \
INPUT=./reads/vars_20x/Sample12.bwa.sam \
OUTPUT=./reads/vars_20x/Sample12.bwa.sort.bam \
SORT_ORDER=coordinate
转录组使用AddOrReplaceReadGroups.jar
java -jar picard.jar AddOrReplaceReadGroups \
I=./star_2pass/ERR188044Aligned.out.sam \
O=./star_2pass/ERR188044_rg_added_sorted.bam \
SO=coordinate \
RGID=ERR188044 \
RGLB=rna \
RGPL=illumina \
RGPU=hiseq \
RGSM=ERR188044
3.4 标记重复
#picard标记重复
java -jar /software/picard-tools-1.119/MarkDuplicates.jar \
MAX_FILE_HANDLES_FOR_READ_ENDS_MAP=8000 \
INPUT=./reads/vars_20x/Sample12.bwa.sort.bam \
OUTPUT=./reads/vars_20x/Sample12.bwa.sort.dedup.bam \
METRICS_FILE=./reads/vars_20x/Sample12.bwa.sort.dedup.metrics \
VALIDATION_STRINGENCY=LENIENT
3.5 对bam文件建立索引
#samtools对bam文件建立索引
/software/samtools-1.3/samtools index \
./reads/vars_20x/Sample12.bwa.sort.dedup.bam
由于 STAR 软件使用的 MAPQ 标准与 GATK 不同,而且比对时会有 reads 的片段落到内含子区间,需要进行一步 MAPQ 同步和 reads 剪切,使用 GATK 专为 RNA-seq 应用开发的工具 SplitNCigarReads 进行操纵,它会将落在内含子区间的 reads 片段直接切除,并对 MAPQ 进行调整。DNA 测序的重测序应用中也有序列比对软件的 MAPQ 与 GATK 无法直接对接的情况,需要进行调整。
java -jar GenomeAnalysisTK.jar -T SplitNCigarReads \
-R ./genome/chrX.fa \
-I ./star_2pass/ERR188044_dedup.bam \
-o ./star_2pass/ERR188044_dedup_split.bam \
-rf ReassignOneMappingQuality \
-RMQF 255 \
-RMQT 60 \
-U ALLOW_N_CIGAR_READS
3.6 重排bam文件(选做)
之后就是可选的 Indel Realignment,对已知的 indel 区域附近的 reads 重新比对,可以稍微提高 indel 检测的真阳性率,如果对于某些物种没有已知indel区域的话官网推荐可以跳过这步直接生成vcf文件,之后使用第一次生成的vcf文件作为已知indel再走一遍流程。
#重排
##第一步首先生成位置信息
java -jar /software/GenomeAnalysisTK.jar \
-R ./ref/TAIR9_chr1.random.fasta \
-T RealignerTargetCreator \
-o ./reads/vars_20x/Sample12.bwa.sort.dedup.realn_known.intervals \
-I ./reads/vars_20x/Sample12.bwa.sort.dedup.bam \
-known ./variants/TAIR9_chr1.random.indel.vcf
##根据上一步位置信息,生成重排好的bam文件
java -jar /software/GenomeAnalysisTK.jar \
-R ./ref/TAIR9_chr1.random.fasta \
-T IndelRealigner \
-targetIntervals ./reads/vars_20x/Sample12.bwa.sort.dedup.realn_known.intervals \
-o ./reads/vars_20x/Sample12.bwa.sort.dedup.realn_known.bam \
-I ./reads/vars_20x/Sample12.bwa.sort.dedup.bam \
-known ./variants/TAIR9_chr1.random.indel.vcf \
--consensusDeterminationModel KNOWNS_ONLY \
-LOD 0.4
3.7 BQSR矫正bam文件(选做)
主要是针对测序质量不是特别高的数据
#bam文件矫正,选做,看测序质量
java -jar /software/GenomeAnalysisTK.jar \
-T BaseRecalibrator \
-R ./ref/TAIR9_chr1.random.fasta \
-I ./reads/vars_20x/Sample12.bwa.sort.dedup.realn_known.bam \
-knownSites ./variants/TAIR9_chr1.random.vcf \
-o ./reads/vars_20x/Sample12.bwa.sort.dedup.realn_known.recal.table
java -jar /software/GenomeAnalysisTK.jar \
-T PrintReads \
-R ./ref/TAIR9_chr1.random.fasta \
-I ./reads/vars_20x/Sample12.bwa.sort.dedup.realn_known.bam \
-BQSR ./reads/vars_20x/Sample12.bwa.sort.dedup.realn_known.recal.table \
-o ./reads/vars_20x/Sample12.bwa.sort.dedup.realn_known.recal.bam
3.8 变异检测,使用HaplotypeCaller
#call variation发现变异并生成gvcf文件
java -jar /software/GenomeAnalysisTK.jar \
-R ./ref/TAIR9_chr1.random.fasta \
-T HaplotypeCaller \
-I ./reads/vars_20x/Sample12.bwa.sort.dedup.realn_known.recal.bam \
-nct 2 \
--emitRefConfidence GVCF \
--variant_index_type LINEAR \
--variant_index_parameter 128000 \
-o ./reads/vars_20x/Sample12.bwa.sort.dedup.realn_known.recal.hc.gvcf
3.9 整合gvcf文件并转化为vcf文件
#整合gvcf文件
java -jar /software/GenomeAnalysisTK.jar \
-R ./ref/TAIR9_chr1.random.fasta \
-T CombineGVCFs \
-o ./variants/Samples/Samples.bwa.sort.dedup.realn_known.recal.hc.gvcf \
-V ./reads/vars_20x/Sample12.bwa.sort.dedup.realn_known.recal.hc.gvcf \
-V ./reads/vars_20x/Sample13.bwa.sort.dedup.realn_known.recal.hc.gvcf
#gvcf文件转化为vcf文件
java -jar /software/GenomeAnalysisTK.jar \
-R ./ref/TAIR9_chr1.random.fasta \
-T GenotypeGVCFs \
-nt 8 \
-V ./variants/Samples/Samples.bwa.sort.dedup.realn_known.recal.hc.gvcf \
-o ./variants/Samples/Samples.bwa.sort.dedup.realn_known.recal.hc.vcf
3.10 VariantRecalibrator校准SNP
java -jar /software/GenomeAnalysisTK.jar \
-T VariantRecalibrator \
-R ./ref/TAIR9_chr1.random.fasta \
-input ./variants/Samples/Samples.bwa.sort.dedup.realn_known.recal.hc.vcf \
-resource:original,known=false,training=true,truth=true,prior=15.0 \
./variants/TAIR9_chr1.random.snp.vcf \
-an DP -an QD -an FS -an MQRankSum -an ReadPosRankSum \
-mode SNP \
--maxGaussians 2 --maxNegativeGaussians 1 --minNumBadVariants 1000 \
-tranche 100.0 -tranche 99.9 -tranche 99.0 -tranche 90.0 \
-recalFile
./variants/Samples/Samples.bwa.sort.dedup.realn_known.recal.hc.SNP.recal \
-tranchesFile
./variants/Samples/Samples.bwa.sort.dedup.realn_known.recal.hc.SNP.tranches\
-rscriptFile ./variants/Samples/Samples.bwa.sort.dedup.realn_known.recal.hc.SNP_plots.R
3.11 最终输出
java -jar /software/GenomeAnalysisTK.jar \
-T ApplyRecalibration \
-R ./ref/TAIR9_chr1.random.fasta \
-input ./variants/Samples/Samples.bwa.sort.dedup.realn_known.recal.hc.vcf \
-mode SNP \
--ts_filter_level 99.0 \
-recalFile ./variants/Samples/Samples.bwa.sort.dedup.realn_known.recal.hc.SNP.recal\
-tranchesFile ./variants/Samples/Samples.bwa.sort.dedup.realn_known.recal.hc.SNP.tranches\
-o ./variants/Samples/Samples.bwa.sort.dedup.realn_known.recal.hc.recal_SNP.vcf
3.12 后续分析
随后可以使用annovar对vcf文件进行注释,可以使用convert2annovar.pl进行格式转换。比如将VCF和pileup格式的文件转换为annovar的输入格式。
#对gtf文件进行注释
/software/UCSC/gtfToGenePred -genePredExt test.gtf test_refGene.txt
#进行注释
convert2annovar.pl -format pileup variant.pileup -outfile variant.query
convert2annovar.pl -format vcf4 test.vcf > test.avinput
#输出注释文件
annotate_variation.pl -hgvs -buildver test_refGene.txt --geneanno test.avinput refdir/
-outfile annotate.snp