--- 大师,大师,我想学习单细胞
··· 闭上眼睛跟我来
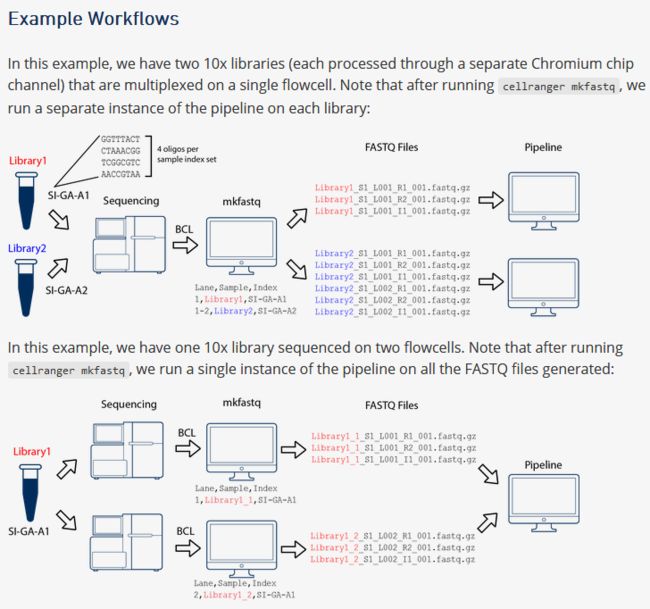
单细胞测序有着漫长的过去,却只有短暂的历史---谁说的!
说她漫长是因为到如今也有十几年的历史了,说她段短暂是因为针对单细胞的分析工具越来越有意义开发周期却越来越短。一般生物信息流程主要由软件(安装与参数)、数据库(结构和生物学意义)和数据分析(统计学和编程)组成,目前单细胞分析用到的软件主要是FastQC、Cellranger和R包Seurat、monocle;数据库有相应物种的参考基因组、KEGG、GO;数据分析部分主要基于count矩阵和差异表达数据用R或者Python来做。
cellranger是10X genomics公司为单细胞RNA测序分析量身打造的数据分析软件,可以直接输入Illumina 原始数据(raw base call ,BCL)输出表达定量矩阵、降维(pca),聚类(Graph-based& K-Means)以及可视化(t-SNE)结果,结合配套的Loupe Cell Browser给予研究者更多探索单细胞数据的机会。cellranger的高度集成化,使得单细胞测序数据探索变得更加简单,研究者有更多的时间来做生物学意义的挖掘。
今天小编就要给大家介绍这下这个可能成为行业标本的数据分析软件——cellranger。在这之前,还是先来了解一下10X genomics单细胞测序的一般原理吧。
10X genomics
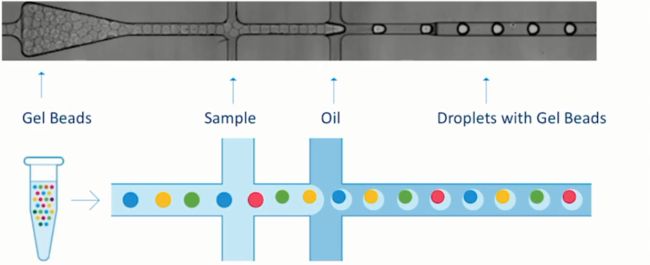
一个油滴=一个单细胞=一个凝胶微珠=一个RNA-Seq,可以说这就是10X的基本技术原理。
V2试剂盒产生的文库结构:
V3试剂盒产生的文库结构:

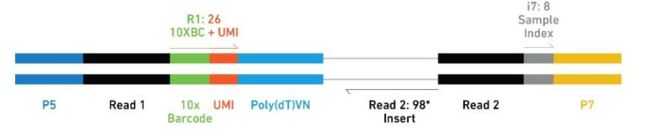
- reads 1 :主要用来定量(barcode、UMI以及reads的来源)
- reads 2 :与基因组比对
- Barcode:标记细胞
- UMI (Unique Molecular Identifier):标记基因
- PolyT :真核生物
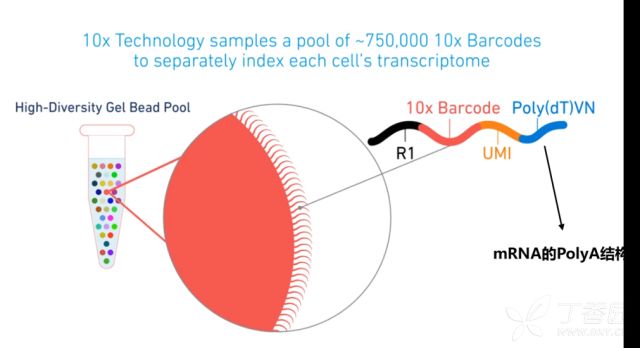
10X genomics单细胞测序通过Barcode来标记细胞,UMI 来标记转录本,这样与参考基因比对后就可以定量细胞以及基因的数量。
在2019年3月7日10x官方网站对“单细胞基因表达入门”的直播视频中提到(只是一场直播提到的信息,仅供参考): 由于10x单细胞测序的重复性较高,因此如无特殊原因,做生物学重复的意义不大;如果细胞大小不一致,但直径符合上机要求,对捕获效率没有明显影响;细胞重悬清洗后要保证钙、镁离子浓度较低,从而不影响反转录;非常规形态或直径较大的细胞可以采用抽核的方法进行检测。当被问及测序时需不需要加入标准物(如ERCC)的时候,官方给出的建议是不建议加ERCC(考虑到成本和影响细胞和基因的计数)。
cell ranger pipeline
cellranger单细胞分析流程主要分为:数据拆分(cellranger mkfastq、细胞定量cellranger count、组合分析cellranger aggr、参数调整cellranger reanalyze。还有一些用户可能会用到的功能:mat2csv、mkgtf、mkref、vdj、mkvdjref、testrun、upload、sitecheck。
本文主要介绍常用的count aggr以及reanlyze。
- cellranger mkfastq
- 拆分
封装了Illumina's bcl2fastq软件,用来拆分Illumina 原始数据(raw base call (BCL)),输出 FASTQ 文件。
cellranger-cs/3.0.0 -h
cellranger mkfastq (3.0.0)
Copyright (c) 2018 10x Genomics, Inc. All rights reserved.
-------------------------------------------------------------------------------
Run Illumina demultiplexer on sample sheets that contain 10x-specific sample
index sets, and generate 10x-specific quality metrics after the demultiplex.
Any bcl2fastq argument will work (except a few that are set by the pipeline
to ensure proper trimming and sample indexing). The FASTQ output generated
will be the same as when running bcl2fastq directly.
These bcl2fastq arguments are overridden by this pipeline:
--fastq-cluster-count
--minimum-trimmed-read-length
--mask-short-adapter-reads
Usage:
cellranger mkfastq --run=PATH [options]
cellranger mkfastq -h | --help | --version
Required:
--run=PATH Path of Illumina BCL run folder.
Optional:
# Sample Sheet
--id=NAME Name of the folder created by mkfastq. If not
supplied, will default to the name of the
flowcell referred to by the --run argument.
--csv=PATH
--samplesheet=PATH
--sample-sheet=PATH Path to the sample sheet. The sample sheet can either
be a simple CSV with lane, sample and index columns,
or an Illumina Experiment Manager-compatible
sample sheet. Sample sheet indexes can refer to
10x sample index set names (e.g., SI-GA-A12).
--simple-csv=PATH Deprecated. Same meaning as --csv.
--ignore-dual-index On a dual-indexed flowcell, ignore the second sample
index, if the second sample index was not used
for the 10x sample.
--qc Calculate both sequencing and 10x-specific metrics,
including per-sample barcode matching rate. Will
not be performed unless this flag is specified.
# bcl2fastq Pass-Through
--lanes=NUMS Comma-delimited series of lanes to demultiplex.
Shortcut for the --tiles argument.
--use-bases-mask=MASK Same as bcl2fastq; override the read lengths as
specified in RunInfo.xml. See Illumina bcl2fastq
documentation for more information.
--delete-undetermined Delete the Undetermined FASTQ files left by
bcl2fastq. Useful if your sample sheet is only
expected to match a subset of the flowcell.
--output-dir=PATH Same as in bcl2fastq. Folder where FASTQs, reports
and stats will be generated.
--project=NAME Custom project name, to override the samplesheet or
to use in conjunction with the --csv argument.
# Martian Runtime
--jobmode=MODE Job manager to use. Valid options:
local (default), sge, lsf, or a .template file
--localcores=NUM Set max cores the pipeline may request at one time.
Only applies when --jobmode=local.
--localmem=NUM Set max GB the pipeline may request at one time.
Only applies when --jobmode=local.
--mempercore=NUM Set max GB each job may use at one time.
Only applies in cluster jobmodes.
--maxjobs=NUM Set max jobs submitted to cluster at one time.
Only applies in cluster jobmodes.
--jobinterval=NUM Set delay between submitting jobs to cluster, in ms.
Only applies in cluster jobmodes.
--overrides=PATH The path to a JSON file that specifies stage-level
overrides for cores and memory. Finer-grained
than --localcores, --mempercore and --localmem.
Consult the 10x support website for an example
override file.
--uiport=PORT Serve web UI at http://localhost:PORT
--disable-ui Do not serve the UI.
--noexit Keep web UI running after pipestance completes or fails.
--nopreflight Skip preflight checks.
-h --help Show this message.
--version Show version.
Note: 'cellranger mkfastq' can be called in two ways, depending on how you
demultiplexed your BCL data into FASTQ files.
1. If you demultiplexed with 'cellranger mkfastq' or directly with
Illumina bcl2fastq, then set --fastqs to the project folder containing
FASTQ files. In addition, set --sample to the name prefixed to the FASTQ
files comprising your sample. For example, if your FASTQs are named:
subject1_S1_L001_R1_001.fastq.gz
then set --sample=subject1
2. If you demultiplexed with 'cellranger demux', then set --fastqs to a
demux output folder containing FASTQ files. Use the --lanes and --indices
options to specify which FASTQ reads comprise your sample.
This method is deprecated. Please use 'cellranger mkfastq' going forward.
有以下两种使用方式
$ cellranger mkfastq --id=tiny-bcl \
--run=/path/to/tiny_bcl \
--csv=cellranger-tiny-bcl-simple-1.2.0.csv
cellranger mkfastq
Copyright (c) 2017 10x Genomics, Inc. All rights reserved.
-------------------------------------------------------------------------------
Martian Runtime - 3.0.2-v3.2.0
Running preflight checks (please wait)...
2017-08-09 16:33:54 [runtime] (ready) ID.tiny-bcl.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET
2017-08-09 16:33:57 [runtime] (split_complete) ID.tiny-bcl.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET
2017-08-09 16:33:57 [runtime] (run:local) ID.tiny-bcl.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET.fork0.chnk0.main
2017-08-09 16:34:00 [runtime] (chunks_complete) ID.tiny-bcl.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET
... # 只有3列,第一列指定lane ID, 第二列指定样本名称,第三列指定index的名称,10X genomics的每个index代表4条具体的oligo序列
$ cellranger mkfastq --id=tiny-bcl \
--run=/path/to/tiny_bcl \
--samplesheet=cellranger-tiny-bcl-samplesheet-1.2.0.csv
cellranger mkfastq
Copyright (c) 2017 10x Genomics, Inc. All rights reserved.
-------------------------------------------------------------------------------
Martian Runtime - 3.0.2-v3.2.0
Running preflight checks (please wait)...
2017-08-09 16:25:49 [runtime] (ready) ID.tiny-bcl.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET
2017-08-09 16:25:52 [runtime] (split_complete) ID.tiny-bcl.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET
2017-08-09 16:25:52 [runtime] (run:local) ID.tiny-bcl.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET.fork0.chnk0.main
2017-08-09 16:25:58 [runtime] (chunks_complete) ID.tiny-bcl.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET
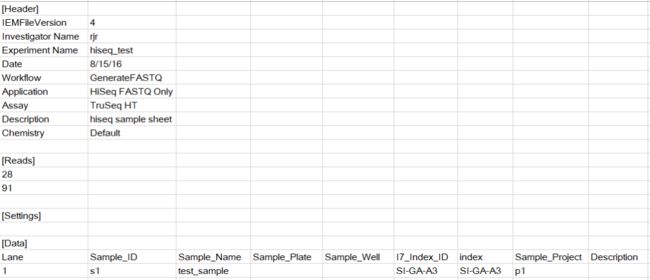
如果使用samplesheet文件,需要调整[Reads]中的序列长度,而使用简化版的csv文件,cell ranger可以识别所用试剂盒版本,然后自动化的调整reads长度。
拆分好之后的目录结构如下所示
├── fastq_path
│ ├── H35KCBCXY
│ │ └── test_sample
│ │ ├── test_sample_S1_L001_I1_001.fastq.gz #index序列
│ │ ├── test_sample_S1_L001_R1_001.fastq.gz
│ │ └── test_sample_S1_L001_R2_001.fastq.gz
- QC 其实这部分并不能叫做Quality control而应该叫做Quality check.
--qc 参数输出序列质量情况,保存在outs/qc_summary.json 中:
"sample_qc": {
"Sample1": {
"5": {
"barcode_exact_match_ratio": 0.9336158258904611,
"barcode_q30_base_ratio": 0.9611993091728814,
"bc_on_whitelist": 0.9447542078230667,
"mean_barcode_qscore": 37.770630795934,
"number_reads": 2748155,
"read1_q30_base_ratio": 0.8947676653366835,
"read2_q30_base_ratio": 0.7771883245304577
},
"all": {
"barcode_exact_match_ratio": 0.9336158258904611,
"barcode_q30_base_ratio": 0.9611993091728814,
"bc_on_whitelist": 0.9447542078230667,
"mean_barcode_qscore": 37.770630795934,
"number_reads": 2748155,
"read1_q30_base_ratio": 0.8947676653366835,
"read2_q30_base_ratio": 0.7771883245304577
}
}
}
cellranger count
count是cellranger最主要也是最重要的功能:完成细胞和基因的定量,也就是产生了我们用来做各种分析的基因表达矩阵。
cellranger count -h
cellranger count (3.0.0)
Copyright (c) 2018 10x Genomics, Inc. All rights reserved.
-------------------------------------------------------------------------------
'cellranger count' quantifies single-cell gene expression.
The commands below should be preceded by 'cellranger':
Usage:
count
--id=ID
[--fastqs=PATH]
[--sample=PREFIX]
--transcriptome=DIR
[options]
count [options]
count -h | --help | --version
Arguments:
id A unique run id, used to name output folder [a-zA-Z0-9_-]+.
fastqs Path of folder created by mkfastq or bcl2fastq.
sample Prefix of the filenames of FASTQs to select.
transcriptome Path of folder containing 10x-compatible reference.
Options:
# Single Cell Gene Expression
--description=TEXT Sample description to embed in output files.
--libraries=CSV CSV file declaring input library data sources.
--expect-cells=NUM Expected number of recovered cells.
--force-cells=NUM Force pipeline to use this number of cells, bypassing
the cell detection algorithm.
--feature-ref=CSV Feature reference CSV file, declaring feature-barcode
constructs and associated barcodes.
--nosecondary Disable secondary analysis, e.g. clustering. Optional.
--r1-length=NUM Hard trim the input Read 1 to this length before
analysis.
--r2-length=NUM Hard trim the input Read 2 to this length before
analysis.
--chemistry=CHEM Assay configuration. NOTE: by default the assay
configuration is detected automatically, which
is the recommened mode. You usually will not need
to specify a chemistry. Options are: 'auto' for
autodetection, 'threeprime' for Single Cell 3',
'fiveprime' for Single Cell 5', 'SC3Pv1' or
'SC3Pv2' or 'SC3Pv3' for Single Cell 3' v1/v2/v3,
'SC5P-PE' or 'SC5P-R2' for Single Cell 5'.
paired-end/R2-only. Default: auto.
--no-libraries Proceed with processing using a --feature-ref but no
feature-barcoding data specified with the
'libraries' flag.
--lanes=NUMS Comma-separated lane numbers.
--indices=INDICES Comma-separated sample index set "SI-001" or sequences.
--project=TEXT Name of the project folder within a mkfastq or
bcl2fastq-generated folder to pick FASTQs from.
# Martian Runtime
--jobmode=MODE Job manager to use. Valid options:
local (default), sge, lsf, or a .template file
--localcores=NUM Set max cores the pipeline may request at one time.
Only applies when --jobmode=local.
--localmem=NUM Set max GB the pipeline may request at one time.
Only applies when --jobmode=local.
--mempercore=NUM Set max GB each job may use at one time.
Only applies in cluster jobmodes.
--maxjobs=NUM Set max jobs submitted to cluster at one time.
Only applies in cluster jobmodes.
--jobinterval=NUM Set delay between submitting jobs to cluster, in ms.
Only applies in cluster jobmodes.
--overrides=PATH The path to a JSON file that specifies stage-level
overrides for cores and memory. Finer-grained
than --localcores, --mempercore and --localmem.
Consult the 10x support website for an example
override file.
--uiport=PORT Serve web UI at http://localhost:PORT
--disable-ui Do not serve the UI.
--noexit Keep web UI running after pipestance completes or fails.
--nopreflight Skip preflight checks.
-h --help Show this message.
--version Show version.
Note: 'cellranger count' can be called in two ways, depending on how you
demultiplexed your BCL data into FASTQ files.
1. If you demultiplexed with 'cellranger mkfastq' or directly with
Illumina bcl2fastq, then set --fastqs to the project folder containing
FASTQ files. In addition, set --sample to the name prefixed to the FASTQ
files comprising your sample. For example, if your FASTQs are named:
subject1_S1_L001_R1_001.fastq.gz
then set --sample=subject1
2. If you demultiplexed with 'cellranger demux', then set --fastqs to a
demux output folder containing FASTQ files. Use the --lanes and --indices
options to specify which FASTQ reads comprise your sample.
This method is deprecated. Please use 'cellranger mkfastq' going forward.
--nosecondary (optional) Add this flag to skip secondary analysis of the feature-barcode matrix (dimensionality reduction, clustering and visualization). Set this if you plan to use cellranger reanalyze or your own custom analysis.
- Output Files
.outs
├── analysis【数据分析文件夹】
│ ├── clustering【聚类,图聚类和k-means聚类】
│ ├── diffexp【差异分析】
│ ├── pca【主成分分析线性降维】
│ └── tsne【非线性降维信息】
├── cloupe.cloupe【Loupe Cell Browser 输入文件】
├── filtered_feature_bc_matrix【过滤掉的barcode信息】
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
├── filtered_feature_bc_matrix.h5【过滤掉的barcode信息HDF5 format】
├── metrics_summary.csv【CSV format数据摘要】
├── molecule_info.h5【UMI信息,aggregate的时候会用到的文件】
├── raw_feature_bc_matrix【原始barcode信息】
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
├── possorted_genome_bam.bam【比对文件】
├── possorted_genome_bam.bam.bai【索引文件】
├── raw_feature_bc_matrix.h5【原始barcode信息HDF5 format】
├── web_summary.html【网页简版报告以及可视化】
└── *_gene_bar.csv_temp【过程文件】
cellranger aggr
When doing large studies involving multiple GEM wells, run cellranger count on FASTQ data from each of the GEM wells individually, and then pool the results using cellranger aggr, as described here
$ cd /home/jdoe/runs
$ cellranger aggr --id=AGG123 \
--csv=AGG123_libraries.csv \
--normalize=mapped
library_id,molecule_h5
LV123,/opt/runs/LV123/outs/molecule_info.h5
LB456,/opt/runs/LB456/outs/molecule_info.h5
LP789,/opt/runs/LP789/outs/molecule_info.h5
library_id,molecule_h5,batch
LV123,/opt/runs/LV123/outs/molecule_info.h5,v2_lib
LB456,/opt/runs/LB456/outs/molecule_info.h5,v3_lib
LP789,/opt/runs/LP789/outs/molecule_info.h5,v3_lib
Understanding GEM Wells
Each GEM well is a physically distinct set of GEM partitions, but draws barcode sequences randomly from the pool of valid barcodes, known as the barcode whitelist. To keep the barcodes unique when aggregating multiple libraries, we append a small integer identifying the GEM well to the barcode nucleotide sequence, and use that nucleotide sequence plus ID as the unique identifier in the feature-barcode matrix. For example, AGACCATTGAGACTTA-1 and AGACCATTGAGACTTA-2 are distinct cell barcodes from different GEM wells, despite having the same barcode nucleotide sequence.
This number, which tells us which GEM well this barcode sequence came from, is called the GEM well suffix. The numbering of the GEM wells will reflect the order that the GEM wells were provided in the Aggregation CSV.
Outputs:
- Aggregation metrics summary HTML: /home/jdoe/runs/AGG123/outs/web_summary.html
- Aggregation metrics summary JSON: /home/jdoe/runs/AGG123/outs/summary.json
- Secondary analysis output CSV: /home/jdoe/runs/AGG123/outs/analysis
- Filtered feature-barcode matrices MEX: /home/jdoe/runs/AGG123/outs/filtered_feature_bc_matrix
- Filtered feature-barcode matrices HDF5: /home/jdoe/runs/AGG123/outs/filtered_feature_bc_matrix.h5
- Unfiltered feature-barcode matrices MEX: /home/jdoe/runs/AGG123/outs/raw_feature_bc_matrix
- Unfiltered feature-barcode matrices HDF5: /home/jdoe/runs/AGG123/outs/raw_feature_bc_matrix.h5
- Unfiltered molecule-level info: /home/jdoe/runs/AGG123/outs/raw_molecules.h5
- Barcodes of cell-containing partitions: /home/jdoe/runs/AGG123/outs/cell_barcodes.csv
- Copy of the input aggregation CSV: /home/jdoe/runs/AGG123/outs/aggregation.csv
- Loupe Cell Browser file: /home/jdoe/runs/AGG123/outs/cloupe.cloupe
cellranger reanalyze
相比于count和aggr,reanalyze接受更多的可选的参数,可以说count和aggr只是属于探索性分析,这里的reanalyze才是真正开始接触故事的真相。
$ cellranger reanalyze -h
cellranger reanalyze (3.0.0)
Copyright (c) 2018 10x Genomics, Inc. All rights reserved.
-------------------------------------------------------------------------------
'cellranger reanalyze' performs secondary analysis (dimensionality
reduction, clustering and differential expression) on a feature-barcode
matrix generated by the 'cellranger count' or 'cellranger aggr' pipelines.
The analysis takes parameter settings from a CSV file. Please see the following
URL for details on the CSV format:
support.10xgenomics.com/single-cell/software
This pipeline does not support multi-genome samples.
The commands below should be preceded by 'cellranger':
Usage:
reanalyze
--id=ID
--matrix=MATRIX_H5
[options]
reanalyze [options]
reanalyze -h | --help | --version
Arguments:
id A unique run id, used to name output folder [a-zA-Z0-9_-]+.
matrix A feature-barcode matrix containing data for one genome. Should
be the filtered version, unless using --force-cells.
Options:
# Analysis
--description=TEXT Sample description to embed in output files.
--params=PARAMS_CSV A CSV file specifying analysis parameters.
Optional.
--barcodes=BARCODE_CSV A CSV file containing a list of cell barcodes to
use for reanalysis, e.g. barcodes exported from
Loupe Cell Browser. Optional.
--genes=GENES_CSV A CSV file containing a list of feature IDs to
use for reanalysis. For gene expression, this
should correspond to the gene_id field in the
reference GTF should be (e.g. ENSG... for
ENSEMBL-based references).
Optional.
--exclude-genes=GENES_CSV A CSV file containing a list of feature IDs to
exclude for reanalysis. For gene expression,
this should correspond to the gene_id field in
the reference GTF (e.g., ENSG... for
ENSEMBL-based references).
The exclusion is applied after --genes.
Optional.
--agg=AGGREGATION_CSV If the input matrix was produced by
'cellranger aggr', you may pass the same
aggregation CSV in order to retain
per-library tag information in the
resulting .cloupe file. This argument is
required to enable chemistry batch correction
Optional.
--force-cells=NUM Force pipeline to use this number of cells,
bypassing the cell detection algorithm.
Optional.
# Martian Runtime
--jobmode=MODE Job manager to use. Valid options:
local (default), sge, lsf, or a .template file
--localcores=NUM Set max cores the pipeline may request at one time.
Only applies when --jobmode=local.
--localmem=NUM Set max GB the pipeline may request at one time.
Only applies when --jobmode=local.
--mempercore=NUM Set max GB each job may use at one time.
Only applies in cluster jobmodes.
--maxjobs=NUM Set max jobs submitted to cluster at one time.
Only applies in cluster jobmodes.
--jobinterval=NUM Set delay between submitting jobs to cluster, in ms.
Only applies in cluster jobmodes.
--overrides=PATH The path to a JSON file that specifies stage-level
overrides for cores and memory. Finer-grained
than --localcores, --mempercore and --localmem.
Consult the 10x support website for an example
override file.
--uiport=PORT Serve web UI at http://localhost:PORT
--disable-ui Do not serve the UI.
--noexit Keep web UI running after pipestance completes or fails.
--nopreflight Skip preflight checks.
-h --help Show this message.
--version Show version.
Note: 'cellranger reanalyze' can be called in two ways, depending on how you
demultiplexed your BCL data into FASTQ files.
1. If you demultiplexed with 'cellranger mkfastq' or directly with
Illumina bcl2fastq, then set --fastqs to the project folder containing
FASTQ files. In addition, set --sample to the name prefixed to the FASTQ
files comprising your sample. For example, if your FASTQs are named:
subject1_S1_L001_R1_001.fastq.gz
then set --sample=subject1
2. If you demultiplexed with 'cellranger demux', then set --fastqs to a
demux output folder containing FASTQ files. Use the --lanes and --indices
options to specify which FASTQ reads comprise your sample.
This method is deprecated. Please use 'cellranger mkfastq' going forward.
$ cd /home/jdoe/runs
$ ls -1 AGG123/outs/*.h5 # verify the input file exists
AGG123/outs/filtered_feature_bc_matrix.h5
AGG123/outs/filtered_molecules.h5
AGG123/outs/raw_feature_bc_matrix.h5
AGG123/outs/raw_molecules.h5
$ cellranger reanalyze --id=AGG123_reanalysis \
--matrix=AGG123/outs/filtered_feature_bc_matrix.h5 \
--params=AGG123_reanalysis.csv
Outputs:
- Secondary analysis output CSV: /home/jdoe/runs/AGG123_reanalysis/outs/analysis_csv
- Secondary analysis web summary: /home/jdoe/runs/AGG123_reanalysis/outs/web_summary.html
- Copy of the input parameter CSV: /home/jdoe/runs/AGG123_reanalysis/outs/params_csv.csv
- Copy of the input aggregation CSV: /home/jdoe/runs/AGG123_reanalysis/outs/aggregation_csv.csv
- Loupe Cell Browser file: /home/jdoe/runs/AGG123_reanalysis/outs/cloupe.cloupe
数据分析概述
Cell Ranger是由10x genomic公司官方提供的专门用于其单细胞转录组数据分析的软件包。Cell Ranger将前面产生的fastq测序数据比对到参考基因组上,然后进行基因表达定量,生成细胞-基因表达矩阵,并基于此进行细胞聚类和差异表达分析。
比对
基因组比对
Cell Ranger使用star比对软件将reads比对到参考基因组上后使用GTF注释文件进行校正,并区分出外显子区、内含子区、基因间区。具体的区分规则(mapping QC)为:至少50% 比对到外显子上reads记为外显子区、将比对到非外显子区且与内含子区有交集的reads记为内含子区,除此之外均为基因间区。
- MAPQ 校正
对于比对到单个外显子位点但同时比对到1个或多个非外显子位点的reads,对外显子位点进行优先排序,并将reads记为带有MAPQ 255的外显子位点。
- 转录组比对
Cell Ranger进一步将外显子reads与参考转录本比对,寻找兼容性。注释到单个基因信息的reads认为是一个特异的转录本,只有注释到转录本的reads才用于UMI计数。
参考基因组目录结构:
├── fasta
│ └── genome.fa
├── genes
│ └── genes.gtf
├── pickle
│ └── genes.pickle
├── README.BEFORE.MODIFYING
├── reference.json
├── star
│ ├── chrLength.txt
│ ├── chrNameLength.txt
│ ├── chrName.txt
│ ├── chrStart.txt
│ ├── exonGeTrInfo.tab
│ ├── exonInfo.tab
│ ├── geneInfo.tab
│ ├── Genome
│ ├── genomeParameters.txt
│ ├── SA
│ ├── SAindex
│ ├── sjdbInfo.txt
│ ├── sjdbList.fromGTF.out.tab
│ ├── sjdbList.out.tab
│ └── transcriptInfo.tab
└── version
细胞计数(cell QC)
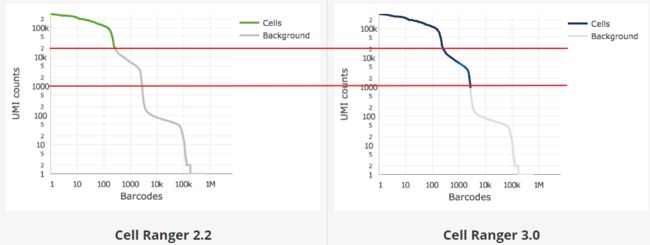
Cell Ranger 3.0引入了一种改进的细胞计数算法,该算法能够更好地识别低RNA含量的细胞群体,特别是当低RNA含量的细胞与高RNA含量的细胞混合时。该算法分为两步:
在第一步中,使用之前的Cell Ranger细胞计数算法识别高RNA含量细胞的主要模式,使用基于每个barcode的UMI总数的cutoff值。Cell Ranger将期望捕获的细胞数量N作为输入(see --expect-cells)。然后将barcodes按照各自的UMI总数由高到低进行排序,取前N个UMI数值的99%分位数为最大估算UMI总数(m),将UMI数目超过m/10的barcodes用于细胞计数。
在第二步中,选择一组具有低UMI计数的barcode,这些barcode可能表示“空的”GEM分区,建立RNA图谱背景模型。利用Simple Good-Turing smoothing平滑算法,对典型空GEM集合中未观测到的基因进行非零模型估计。最后,将第一步中未作为细胞计数的barcode RNA图谱与背景模型进行比较,其RNA谱与背景模型存在较大差异的barcode用于细胞计数。
多态率估计(Estimating Multiplet Rates)
当有多个参考基因组(如人H和鼠M)时,Cell Ranger可以通过多基因组分析区分多物种混合建库的样品,主要根据barcode内每个物种对应的UMI数量进行区分,将其分成H和M两类。最后还会根据H,M各自UMI的分布和最大似然估计法估计多细胞比例(multiplet rate),即(H,M)、(H,H)、(M,M)三种类型的多细胞占比。
基因表达分析(Secondary Analysis of Gene Expression)
尽管我们的表达矩阵经过重重QC,但是单细胞高通量的数据还是表现出高纬度、稀疏性、非正态分布的特点,每一点都是对传统数据分析方法的挑战。于是越来越多的新方法被开发出来,主要借鉴多元分析和机器学习等传统生物统计教材很少教授的方法。越来越多的机器学习方法应用的高通量数据分析中来,那么我们就需要了解机器学习的三个要素;
公式(方法) = 模型 (目的) + 策略(评价) + 算法(实现)
- 降维分析(Dimensionality Reduction)
流形学习(manifold learning)是机器学习、模式识别中的一种方法,在维数约简方面具有广泛的应用。它的主要思想是将高维的数据映射到低维,使该低维的数据能够反映原高维数据的某些本质结构特征。流形学习的前提是有一种假设,即某些高维数据,实际是一种低维的流形结构嵌入在高维空间中。流形学习的目的是将其映射回低维空间中,揭示其本质。
针对单细胞测速数据特点,一般为了提取基因表达矩阵最重要的特征采用降维分析将多维数据的差异投影在低维度上,进而揭示复杂数据背后的规律。Cell Ranger先采用基于IRLBA算法的主成分分析(Principal Components Analysis,PCA)将数据集的维数从(Cell x genes)改变为(Cell x M,M是主成分数量)。然后采用非线性降维算法t-SNE将降维后的数据展示在2维或三维空间中,细胞之间的基因表达模式越相似,在t-SNE图中的距离也越接近。
- 聚类分析(Clustering)
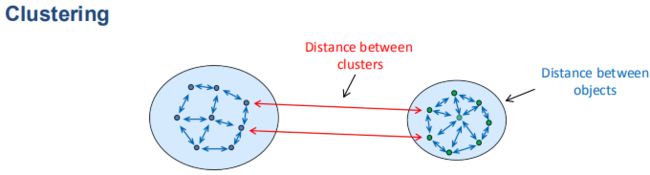
聚类是把相似的对象通过静态分类的方法分成不同的组别或者更多的不连续子集(subset),这样让在同一个子集中的成员对象都有相似的一些属性。在单细胞研究中,聚类分析是识别细胞异质性(heterogeneity)常用的算法。

在PCA空间中,Cell Ranger分别采用Graph-based和K-Means两种不同的聚类方法对细胞进行聚类:
- Graph-based
图聚类算法包括两步:首先用PCA降维的数据构建一个细胞间的k近邻稀疏矩阵,即将一个细胞与其欧式距离上最近的k个细胞聚为一类,然后在此基础上用Louvain算法进行模块优化(Blondel, Guillaume, Lambiotte, & Lefebvre, 2008),旨在找到图中高度连接的模块。最后通过层次聚类将位于同一区域内没有差异表达基因(B-H adjusted p-value 低于0.05)的cluster进一步融合,重复该过程直到没有clusters可以合并。
- k-means
K-Means聚类是无监督的机器学习算法。在PCA降维的空间中随机选取的k个初始质心点,将每个点划分到最近的质心,形成K个簇 ,然后对于每一个cluster重新计算质心并再次进行划分,重复该过程直到收敛。与图聚类算法的k意义不同,这里的k是事先给定的亚群数目。
差异分析(Differential Expression)
为了找到不同细胞亚群之间的差异基因,Cell Ranger使用改进的sSeq方法(基于负二项检验; Yu, Huber, & Vitek, 2013)。当UMI counts数值较大时,为加快分析速度,Cell Ranger会自动切换到edgeR进行beta检验(Robinson & Smyth, 2007)。通过样品的一个亚群与该样品的所有其他亚群进行比较,得到该亚群细胞与其他亚群细胞之间的差异基因列表。
Cell Ranger's implementation differs slightly from that in the paper: in the sSeq paper, the authors recommend using DESeq's geometric mean-based definition of library size. Cell Ranger instead computes relative library size as the total UMI counts for each cell divided by the median UMI counts per cell. As with sSeq, normalization is implicit in that the per-cell library-size parameter is incorporated as a factor in the exact-test probability calculations.
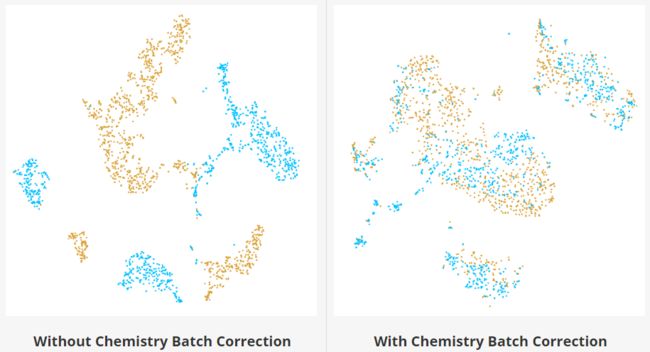
化学批次校正(Chemistry Batch Correction)
为了校正V2V3化学试剂之间的批次效应,Cell Ranger采用一种基于mutual nearest neighbors (MNN;(Haghverdi et al, 2018)的算法来识别批次之间的相似细胞亚群。MNN定义为来自彼此最近邻集合中包含的两个不同批次的细胞群。使用批次之间匹配的细胞亚群,将多个批次合并在一起(Hie et al, 2018)。MNN对中细胞间表达值的差异提供了批次效应的估计。每个细胞校正向量的加权平均值用来估计批次效应。
批次效应得分(batch effect score )被定义为定量测量校正前后的批次效应。对于每个细胞,计算其k个最近的细胞(nearest-neighbors)中有m个属于同一批次,并在没有批次效应时将其标准化为相同批次细胞的期望值M。批次效应得分计算为随机抽取10%的细胞总数S中的上述度量的平均值。如果没有批次效应,我们可以预期每个单元格的最近邻居将在所有批次中均匀共享,批次效应得分接近1。
最近单细胞的小伙伴都在看的一篇文章,不知道你看了没:单细胞测序(scRNA-seq)数据处理必知必会
全文完
cellranger-3.0.0/cellranger-cs/3.0.0/bin
cellranger (3.0.0)
Copyright (c) 2018 10x Genomics, Inc. All rights reserved.
-------------------------------------------------------------------------------
Usage:
cellranger mkfastq
cellranger count
cellranger aggr
cellranger reanalyze
cellranger mat2csv
cellranger mkgtf
cellranger mkref
cellranger vdj
cellranger mkvdjref
cellranger testrun
cellranger upload
cellranger sitecheck
What is Cell Ranger?
使用cell ranger拆分10X单细胞转录组原始数据
使用cell ranger进行单细胞转录组定量分析
专门分析10x genomic公司的单细胞转录组数据的软件套件
10X Genomics 单细胞 RNA-Seq
10X genomics单细胞数据集探索
cellranger
System Requirements
Getting started with Cell Ranger
Cell Ranger 2.1.0 Gene Expression
CG000201_TechNote_Chromium_Single_Cell_3___v3_Reagent__Workflow___Software_Updates_RevA (1)
bcl2fastq Conversion
实验记录2:Cellranger count整理序列数据
Single-Library Analysis with Cell Ranger
Aggregating Multiple GEM Groups with cellranger aggr
单个细胞的测序?(part 2)
PIPELINES
Gene Expression Algorithms Overview
单细胞(single cell)分选平台比较(10X Genomics,BD Rhapsody)
mnnCorrect: Mutual nearest neighbors correction
Gephi网络图极简教程