该教程来自ampvis2官方网站。
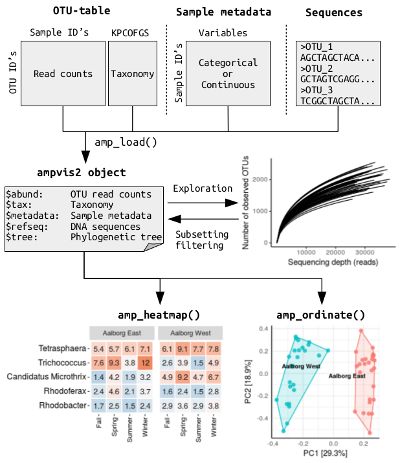
1. ampvis2包的工作流程
2. ampvis2包安装
我是使用的Windows10系统下的R 3.5.3和RStudio,同时需要安装RTools。打开Rstudio后运行以下代码进行安装:
> install.packages("remotes")
> remotes::install_github("MadsAlbertsen/ampvis2")
3. 载入数据
ampvis2能够接受常用的扩增子分析流程 QIIME, mothur以及 UPARSE产出的结果文件。 QIIME和mothur (通过 make.biom 代码) 都能以BIOM文件格式导出数据结果,然后通过ampvis2包中的amp_import_biom功能将数据导入到R中。UPARSE产出的数据可以通过ampvis2包中的 amp_import_uparse功能将数据导入到R中。 示例数据为minimal example data。
#下载示例数据解压后有两个csv文件,设置一个ampvis2_test工作目录,将csv文件放到该目录下。
> setwd("C:/Users/Administrator/Desktop/R_work/ampvis2_test")
> list.files()
[1] "example_metadata.csv" "example_otutable.csv"
> myotutable <- read.csv("example_otutable.csv", check.names = FALSE)
> mymetadata <- read.csv("example_metadata.csv", check.names = FALSE)
> library(ampvis2)
> d <- amp_load(otutable = myotutable,
metadata = mymetadata)
4. 数据过滤和子数据集构建
ampvis2包中包含有一个大的示例数据集MiDAS,这个数据集是在2006-2013年期间从污水处理植物后取样573个活性污泥的扩增子样品。
> data("MiDAS")
> MiDAS
ampvis2 object with 5 elements.
Summary of OTU table:
Samples OTUs Total#Reads Min#Reads Max#Reads Median#Reads Avg#Reads
658 14969 20890850 10480 46264 31800 31749.01
Assigned taxonomy:
Kingdom Phylum Class Order Family Genus Species
14969(100%) 14477(96.71%) 12737(85.09%) 11470(76.63%) 9841(65.74%) 7380(49.3%) 28(0.19%)
Metadata variables: 5
SampleID, Plant, Date, Year, Period
#查看数据集中的序列
> MiDAS$refseq
14969 DNA sequences in binary format stored in a list.
Mean sequence length: 472.922
Shortest sequence: 425
Longest sequence: 525
Labels:
OTU_1
OTU_2
OTU_3
OTU_4
OTU_5
OTU_6
...
Base composition:
a c g t
0.261 0.225 0.319 0.194
(Total: 7.08 Mb)
#从MiDAS中选取plant变量中的Aalborg West和Aalborg East数据作为子数据集
> MiDASsubset <- amp_subset_samples(MiDAS, Plant %in% c("Aalborg West", "Aalborg East"))
590 samples and 5512 OTUs have been filtered
Before: 658 samples and 14969 OTUs
After: 68 samples and 9457 OTUs
#或者选取更为复杂的子数据集
> MiDASsubset <- amp_subset_samples(MiDAS, Plant %in% c("Aalborg West", "Aalborg East") & !SampleID %in% c("16SAMP-749"), minreads = 10000)
591 samples and 5539 OTUs have been filtered
Before: 658 samples and 14969 OTUs
After: 67 samples and 9430 OTUs
5. 热图绘制
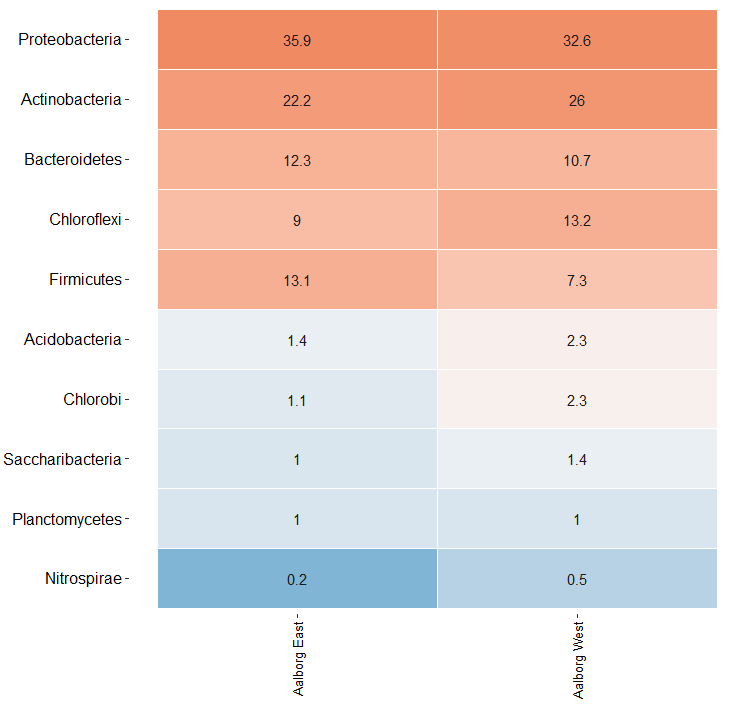
简单热图,只展示了Aalborg West和Aalborg East两个不同实验组中相对丰度最高的10个门水平的微生物群落。
> amp_heatmap(MiDASsubset, group_by = "Plant")
复杂热图, 展示了两个实验组中样品取样时间,相对丰度最高的25个属水平的微生物群落并且标明了对应的门分类水平,相对丰度高低的图例等信息。
> amp_heatmap(MiDASsubset,
group_by = "Plant",
facet_by = "Year",
tax_aggregate = "Genus",
tax_add = "Phylum",
tax_show = 25,
color_vector = c("white", "darkred"),
plot_colorscale = "sqrt",
plot_values = FALSE) +
theme(axis.text.x = element_text(angle = 45, size=10, vjust = 1),
axis.text.y = element_text(size=8),
legend.position="right")

6. 箱线图绘制
简单箱线图,子数据集中所有样品的属水平微生物群落按照相对丰度均值高低排序。
> amp_boxplot(MiDASsubset)

复杂箱线图,根据不同的采用季节分组显示,添加属分类水平群落对应的门分类水平信息。
> amp_boxplot(MiDASsubset,
group_by = "Period",
tax_show = 5,
tax_add = "Phylum")

7. 排序分析Ordination analysis
基于Bray-Curtis距离矩阵的主坐标分析PCoA
> amp_ordinate(MiDASsubset,
type = "pcoa",
distmeasure = "bray",
sample_color_by = "Plant",
sample_colorframe = TRUE,
sample_colorframe_label = "Plant") + theme(legend.position = "blank")

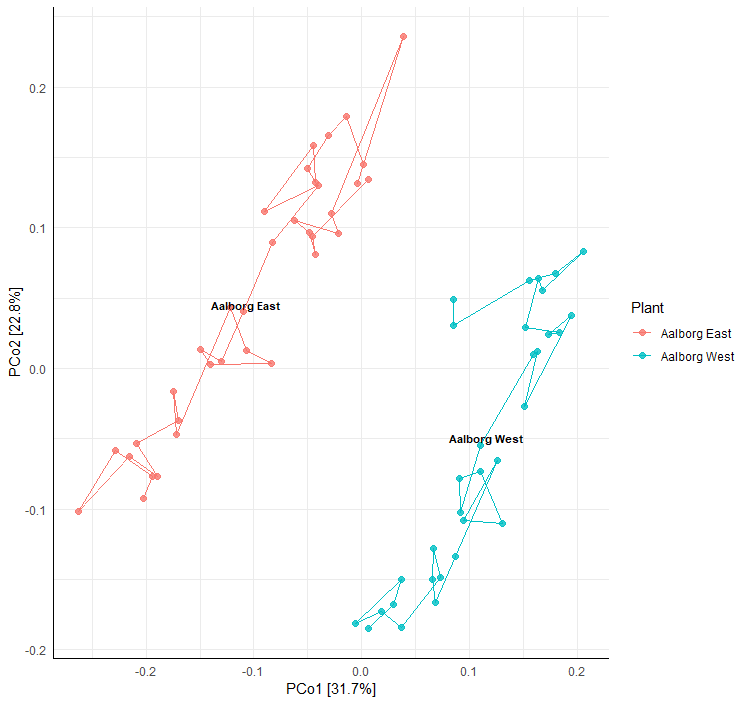
添加取样时间信息,点之间的连线标明取样时间轨迹。
> amp_ordinate(MiDASsubset,
type = "pcoa",
distmeasure = "bray",
sample_color_by = "Plant",
sample_colorframe_label = "Plant",
sample_trajectory = "Date",
sample_trajectory_group = "Plant")
典型对应分析CCA,展示了样品不同季节的分布情况。
> ordinationresult <- amp_ordinate(MiDASsubset,
type = "CCA",
constrain = "Period",
transform = "Hellinger",
sample_color_by = "Period",
sample_shape_by = "Plant",
sample_colorframe = TRUE,
sample_colorframe_label = "Period",
detailed_output = TRUE)
> ordinationresult$plot

此外该R包还能其他分析作图功能,需要自己去探索。
amp_rarecurve()
amp_octave()
amp_timeseries()
amp_venn()
amp_core()
amp_frequency()
amp_otu_network()
amp_rankabundance()
amp_alphadiv()