- JVM常用概念之FPU溢出
剑海风云
JDK(JavaDevelopmentKit)jvmFPU溢出
问题当自己的代码根本没有浮点或矢量运算,JVM在x86生成的机器代码为什么会用到XMM寄存器?基础知识FPU和矢量单元在现代CPU中随处可见,在许多情况下,它们为FPU特定的操作提供了一组备用寄存器。例如,Intelx86_64中的SSE和AVX扩展具有一组额外的宽XMM、YMM和ZMM寄存器,可与更宽的指令结合使用。虽然非矢量指令集通常与矢量和非矢量寄存器不正交(例如,我们不能在x86_64上将
- JVM常用概念之安全点
剑海风云
JDK(JavaDevelopmentKit)jvm安全点mutator线程
1.什么是安全点?安全点是执行线程状态被充分描述的执行范围。安全点是常见的JVM实现细节;在安全点处,mutator线程处于与堆交互的已知且定义明确的点。这意味着堆栈上的所有引用都已映射(在已知位置),并且JVM可以对所有引用进行解释。只要线程保持在安全点处,我们就可以安全地操作堆+堆栈,这样当线程离开安全点时,它对世界的视图就保持一致。目前所有的JVM都对全局安全点有一定的要求如果Java线程被
- C51芯片包下载安装
Book_熬夜!
环境配置有关51单片机
一、前言由于前段时间下载Keil5用于编写stm32单片机程序,最近需要编写C51单片机的程序,在创建新项目时发现没有51单片机的器件型号,花了一点时间解决这个问题,故在此分享。二、解决方法1、下载烧录软件stc-isp百度网盘链接提取码:spvx解压密码:51打开后选择使用的芯片->Keil仿真设计->添加型号和头文件到Keil中即可。2、常见报错在执行上述操作时,注意添加时要求Keil安装的一
- 数据结构与算法——二叉树,多叉树的递归遍历、层序遍历,DFS与BFS
Book_熬夜!
数据结构与算法深度优先宽度优先算法数据结构广度优先
文章目录二叉树1.递归遍历2.层序遍历3.多叉树遍历二叉树【子节点】:每个节点下方相连的节点【父节点】:每个节点上方相连的节点【根节点】:最上方没有父节点的节点【叶子节点】:最下方没有子节点的节点【最大深度】:树的最大层数【高度】:节点数减一,即枝数。【满二叉树(PerfectBinaryTree)】:深度为h,则总节点数:2^h-1FullBinaryTree是指一棵二叉树的所有节点要么没有孩子
- 数据结构——环形数组
Book_熬夜!
数据结构与算法数据结构javascript算法
环形数组start指向第一个有效元素的索引,end指向最后一个有效元素的下一个位置索引。注意:start是闭区间,先左移后赋值,先赋值(null)后右移;end是开区间,先赋值再右移,先左移再赋值(null)。左移减一加size再取模,右移加一再取模。【JS代码实现:】classCycleArray{constructor(size=1){this.size=size;this.arr=newAr
- Spring Boot整合SA-Token的使用详解
陈辰学长
springboot数据库后端
SpringBoot整合SA-Token的使用详解,涉及到SA-Token的基本介绍、整合步骤、配置、常用API以及实际使用场景等多个方面。以下将详细阐述这一过程,确保内容不少于2000字。一、SA-Token简介SA-Token是一个轻量级的Java权限认证框架,由国人开发,主要解决登录认证、权限认证、单点登录、OAuth2.0、分布式Session会话、微服务网关鉴权等一系列权限相关问题。SA
- nginx中proxy_pass和root的区别
LeonNo11
nginxnginx运维
在location配置中,proxy_pass和root是完全不同的指令,它们的作用和适用场景不同。1.proxy_pass:代理请求到后端服务器location/api/{proxy_passhttp://http_backend;}作用把请求转发到后端服务器http_backend,即Nginx作为反向代理。适用于Nginx作为API网关或负载均衡的情况。示例如果http_backend是ht
- 使用Python Flask构建Web应用程序
代码快速拳
pythonflask前端Python
Flask是一个轻量级的PythonWeb框架,它提供了构建Web应用程序所需的基本功能。它简单易用,非常适合小型项目和原型开发。本文将介绍如何使用Flask构建一个简单的Web应用程序,并提供相应的源代码。首先,我们需要安装Flask。可以使用以下命令使用pip安装Flask:pipinstallflask一旦安装完成,我们就可以开始构建我们的Web应用程序了。首先,创建一个Python文件,命
- 2024年一文1800字从0到1使用Python Flask实战构建Web应用(1)
2401_84564025
程序员pythonflask前端
现在我也找了很多测试的朋友,做了一个分享技术的交流群,共享了很多我们收集的技术文档和视频教程。如果你不想再体验自学时找不到资源,没人解答问题,坚持几天便放弃的感受可以加入我们一起交流。而且还有很多在自动化,性能,安全,测试开发等等方面有一定建树的技术大牛分享他们的经验,还会分享很多直播讲座和技术沙龙可以免费学习!划重点!开源的!!!qq群号:110685036第三部分:运行Flask应用在app.
- Go语言的数据结构
2401_90032081
包罗万象golang开发语言后端
Go语言的数据结构Go语言(也称为Golang)是一种由谷歌开发的开源编程语言,以其简单性、高效性和并发性而受到欢迎。作为一门现代语言,Go语言在处理数据时提供了丰富的数据结构,这些数据结构不仅可以帮助开发者管理复杂的数据关系,还能提高程序的性能和可读性。本文将详细探讨Go语言中的各种数据结构,包括数组、切片、映射、链表、树以及它们的使用场景与实现细节。一、数组1.1数组的定义在Go语言中,数组是
- OpenStack Heat模板实战:快速创建用户、容器、网络与云主机类型
冯·诺依曼的
openstack网络ssh运维云计算
Heat是OpenStack中的编排服务,通过YAML模板自动化资源管理。本文通过4个实战案例,详解如何用Heat模板创建用户体系、Swift容器、网络资源及云主机类型。一、创建用户、Domain、租户及用户绑定目标:在chinaskillsDomain下创建beijing_group租户,并创建用户cloud。#user_create.ymlheat_template_version:2016-
- Logo语言的学习路线
滕若岚
包罗万象golang开发语言后端
学习Logo语言的路线图引言在计算机编程领域,有许多种编程语言可以选择,Logo语言因其独特的教育理念和简单性而受到广泛欢迎。Logo语言的设计初衷是为了给学生和初学者提供一个轻松愉快的编程学习体验,让他们在学习编程的过程中培养逻辑思维能力和创造力。本文将为您提供一条系统的Logo学习路线,使您能够从基础知识起步,逐渐掌握这门语言。一、Logo语言基础1.1什么是Logo语言?Logo语言最早是在
- Vue 路由中 `routes` 配置项各个属性的详细讲解
遇见~未来
Vue.jsvue.js前端javascript
1.path说明:字符串,表示路由的路径。细节:路径可以包含动态段,例如/user/:id,其中:id是一个动态参数,用于捕获URL中的值并传递给组件。支持通配符,如*,用于匹配所有路径。路径可以是绝对路径(以/开头)或相对路径(不以/开头,通常用于子路由)。示例:{path:'/home',component:Home}{path:'/user/:id',component:User}{path
- 美团Leaf分布式ID生成器:使用详解与核心原理解析
Cloud_.
分布式
引言在分布式系统中,全局唯一ID是贯穿整个业务链路的关键标识,无论是订单号、用户ID、支付流水号,还是日志追踪,都需要唯一且有序的ID来保证数据的一致性。然而,传统的自增ID方案(如数据库自增主键)在分布式场景下面临单点故障、性能瓶颈、分库分表冲突等问题。美团开源的Leaf分布式ID生成器通过创新的设计解决了这些难题,成为业界广泛使用的解决方案之一。本文将深入解析Leaf的两种核心模式(号段模式与
- Vue 路由 (vue-router) 详细总结
遇见~未来
Vue.jsvue.js前端javascript
一、传统web应用与单页面web应用1.1传统web应用传统web应用由多个HTML页面组成,页面切换时会重新加载整个页面,导致用户体验不够流畅,对服务器压力较大。1.2单页面web应用(SPA)单页面应用只有一个HTML页面,通过JavaScript动态更新页面内容,实现局部刷新,具有以下特点:用户体验好:响应性强,类似桌面应用的即时性。服务器压力小:服务器只需提供数据,不负责页面渲染。前后端分
- 全面了解 Vue 路由中 path属性的使用方法和相关配置
遇见~未来
Vue.jsvue.js前端javascript
在Vue路由中,path是一个非常重要的属性,它定义了路由的路径。以下是关于path的详细介绍:1.基本写法path是一个字符串,表示路由的路径。它可以是绝对路径(以/开头)或相对路径(不以/开头,通常用于子路由)。2.是否必须以/开头绝对路径:通常以/开头,表示从根路径开始匹配。例如,/home、/user/profile。相对路径:不以/开头,通常用于子路由,表示相对于父路由的路径。例如,在一
- 利用pprof对golang进行性能分析
忍界英雄
go学习笔记golang
利用pprof进行性能分析pprof性能分析的5个方面一、性能分析的五个核心维度CPU分析-剖析程序的CPU使用情况,定位高耗时函数内存分析-追踪内存分配与泄露,优化内存使用模式IO分析-监控文件/网络IO操作,发现瓶颈资源Goroutine分析-检测协程泄露与异常堆栈并发问题分析-诊断死锁及通过racedetector检测数据竞争数据采集时间生产环境采集:选择业务低峰期进行采样(凌晨2-4点)测
- Java面试系列-ElasticSearch面试题20道,文档,索引,搜索,聚合,分词器,集群管理,索引模版,数据备份和恢复,安全机制,集群扩展,实时搜索,索引生命周期,节点发现,批量操作,基本架构
图苑
java面试elasticsearch
文章目录1.Elasticsearch的基本架构是什么?2.Elasticsearch中的Shard和Replica是如何工作的?3.Elasticsearch中的文档是如何存储的?4.Elasticsearch中的索引是如何创建的?5.Elasticsearch中的搜索是如何工作的?6.Elasticsearch中的聚合是如何工作的?7.Elasticsearch中的分词器是如何工作的?8.El
- 数据结构与算法——二叉搜索树,使用TreeMap将键值对存储在一棵二叉搜索树的节点
Book_熬夜!
数据结构与算法算法javascript数据结构
二叉搜索树【二叉搜索树(BST)】:对于树中的每个节点,其左子树的每个节点的值都要小于这个节点的值,右子树的每个节点的值都要大于这个节点的值。左小右大。中序遍历结果是有序的,会从小到大排序。7/\49/\\1810(不符合)可以使用TreeMap把键值对存储在一棵二叉搜索树的节点里通过遍历这棵二叉搜索树,比遍历普通的二叉树能更快实现增删查改classTreeNode{constructor(key
- 【人工智能基础2】Tramsformer架构、自然语言处理基础、计算机视觉总结
roman_日积跬步-终至千里
人工智能习题人工智能自然语言处理计算机视觉
文章目录七、Transformer架构1.替代LSTM的原因2.Transformer架构:编码器-解码器架构3.Transformer架构原理八、自然语言处理基础1.语言模型基本概念2.向量语义3.预训练语言模型的基本原理与方法4.DeepSeek基本原理九、计算机视觉七、Transformer架构1.替代LSTM的原因处理极长序列时,效率下降:虽然LSTM设计的初衷是解决长期依赖问题,即让模型
- 【python web】一文掌握 Flask 的基础用法
数据知道
python前端flask
文章目录一、Flask介绍1.1安装Flask二、Flask的基本使用2.1创建第一个Flask应用2.2路由与视图函数2.3请求与响应2.4响应对象2.5模板渲染2.6模板继承2.7静态文件管理2.8Blueprint蓝图2.9错误处理三、Flask扩展与插件四、部署Flask应用五、总结Flask是一个轻量级的PythonWeb框架,因其简单易用、灵活性高而受到广泛欢迎。本文将全面介绍Flas
- C# WPF面试题:WPF中一些常见的设计模式
令狐掌门
WPF面试题wpfWPF中的设计模式
C#WPF(WindowsPresentationFoundation)是一个用于创建桌面应用程序的框架,它广泛使用了多种设计模式。以下是一些常见的设计模式:MVVM(Model-View-ViewModel):这是WPF最常用的设计模式。它将数据模型(Model)、视图(View)和视图模型(ViewModel)分离,使得各部分可以独立进行开发和测试。视图模型是视图的抽象,它包含了视图的状态和行
- K8S学习之基础三十四:K8S之监控Prometheus部署pod版
云上艺旅
K8S学习kubernetes学习prometheus云原生
使用KubernetesPod的方式部署Prometheus是一种常见的方法,尤其是在容器化和微服务架构中。以下是详细的步骤:1.创建命名空间(可选)为了方便管理,可以为Prometheus创建一个单独的命名空间。yaml复制apiVersion:v1kind:Namespacemetadata:name:monitoring将上述内容保存为namespace.yaml,然后应用:bash复制ku
- K8S学习之基础三十五:k8s之Prometheus部署模式
云上艺旅
K8S学习kubernetes学习prometheus云原生容器
Prometheus有多种部署模式,适用于不同的场景和需求。以下是几种常见的部署模式:1.单节点部署这是最简单的部署模式,适用于小型环境或测试环境。特点:单个Prometheus实例负责所有的数据采集、存储和查询。配置简单,易于维护。不具备高可用性和扩展性。适用场景:小型项目或测试环境。对高可用性要求不高的场景。部署步骤:下载并解压Prometheus。配置prometheus.yml。启动Pro
- 「C语言指针函数与函数指针:从内存管理到灵活调用的实战指南」
℡残城碎梦
c语言指针函数函数指针函数指针数组
1.指针函数:外卖柜的「生存法则」核心痛点:返回局部变量地址导致崩溃?堆区与栈区傻傻分不清?生活类比:栈区≈临时摊位(函数结束即销毁)堆区≈智能外卖柜(手动申请释放,长期有效)代码对比://错误!返回栈区地址(临时摊位被拆)char*bug_demo(){charbuf[32]="hello";returnbuf;//危险操作!}//正确!返回堆区地址(外卖柜长期存餐)char*correct_d
- Linux----网络tcp编程
weixin_51790712
linux网络tcp/ip
网络编程编程linux操作系统[用户空间]应用层//程序员实现------------------------------------------------------[内核空间]传输层[网络协议栈]//内核已经实现好的属于网络功能网络层数据链路层物理层程序发送数据系统调用---通过系统调用来使用操作系统提供的网络功能函数接口---socketsocket:1.操作系统提供的函数接口//通过这个
- go面试必问,什么是中间件?
走,我们去吹风
中间件golang面试服务器后端
中间件用过么?Middleware是Web的重要组成部分,中间件(通常)是一小段代码,它们接受一个请求,对其进行处理,每个中间件只处理一件事情,完成后将其传递给另一个中间件或最终处理程序,这样就做到了程序的解耦。全局中间件所有的请求都要经过此中间件packagemainimport("fmt""time""github.com/gin-gonic/gin")//定义中间funcMiddleWare
- 自我学习: Django-用户登录+中间件
yzybang
django中间件学习
以form来做,因为form没有写入能力,比较安全fromdjango.shortcutsimportrender,HttpResponse,redirectfromapp01importmodelsfromdjangoimportformsfromapp01.utils.encryptimportmd5#form需自己定义“字段”classLoginForm(forms.Form):name=f
- 解决 HTTP 请求中的编码问题:从乱码到正确传输
和烨
Java进阶学习专栏http网络协议网络
文章目录解决HTTP请求中的编码问题:从乱码到正确传输1.**问题背景**2.**乱码问题的原因**2.1**客户端编码问题**2.2**请求头缺失**2.3**服务器编码问题**3.**解决方案**3.1**明确指定请求体编码**3.2**确保请求头正确**3.3**动态获取响应编码**4.**调试与验证**4.1**打印请求数据**4.2**使用抓包工具**4.3**查看服务器日志**5.**
- java实现大文件传输
M_Snow
java开发语言
简介在现代互联网中,我们经常需要传输大文件,例如视频、音频或者大型数据文件。传输大文件需要考虑诸多因素,例如网络延迟、带宽限制和传输安全性。在本文中,我们将介绍如何使用Java实现大文件传输,并提供相应的代码示例。文件传输协议在进行大文件传输之前,我们需要选择合适的传输协议。目前常用的文件传输协议有FTP(FileTransferProtocol)、SFTP(SecureFileTransferP
- Java开发中,spring mvc 的线程怎么调用?
小麦麦子
springmvc
今天逛知乎,看到最近很多人都在问spring mvc 的线程http://www.maiziedu.com/course/java/ 的启动问题,觉得挺有意思的,那哥们儿问的也听仔细,下面的回答也很详尽,分享出来,希望遇对遇到类似问题的Java开发程序猿有所帮助。
问题:
在用spring mvc架构的网站上,设一线程在虚拟机启动时运行,线程里有一全局
- maven依赖范围
bitcarter
maven
1.test 测试的时候才会依赖,编译和打包不依赖,如junit不被打包
2.compile 只有编译和打包时才会依赖
3.provided 编译和测试的时候依赖,打包不依赖,如:tomcat的一些公用jar包
4.runtime 运行时依赖,编译不依赖
5.默认compile
依赖范围compile是支持传递的,test不支持传递
1.传递的意思是项目A,引用
- Jaxb org.xml.sax.saxparseexception : premature end of file
darrenzhu
xmlprematureJAXB
如果在使用JAXB把xml文件unmarshal成vo(XSD自动生成的vo)时碰到如下错误:
org.xml.sax.saxparseexception : premature end of file
很有可能时你直接读取文件为inputstream,然后将inputstream作为构建unmarshal需要的source参数。InputSource inputSource = new In
- CSS Specificity
周凡杨
html权重Specificitycss
有时候对于页面元素设置了样式,可为什么页面的显示没有匹配上呢? because specificity
CSS 的选择符是有权重的,当不同的选择符的样式设置有冲突时,浏览器会采用权重高的选择符设置的样式。
规则:
HTML标签的权重是1
Class 的权重是10
Id 的权重是100
- java与servlet
g21121
servlet
servlet 搞java web开发的人一定不会陌生,而且大家还会时常用到它。
下面是java官方网站上对servlet的介绍: java官网对于servlet的解释 写道
Java Servlet Technology Overview Servlets are the Java platform technology of choice for extending and enha
- eclipse中安装maven插件
510888780
eclipsemaven
1.首先去官网下载 Maven:
http://www.apache.org/dyn/closer.cgi/maven/binaries/apache-maven-3.2.3-bin.tar.gz
下载完成之后将其解压,
我将解压后的文件夹:apache-maven-3.2.3,
并将它放在 D:\tools目录下,
即 maven 最终的路径是:D:\tools\apache-mave
- jpa@OneToOne关联关系
布衣凌宇
jpa
Nruser里的pruserid关联到Pruser的主键id,实现对一个表的增删改,另一个表的数据随之增删改。
Nruser实体类
//*****************************************************************
@Entity
@Table(name="nruser")
@DynamicInsert @Dynam
- 我的spring学习笔记11-Spring中关于声明式事务的配置
aijuans
spring事务配置
这两天学到事务管理这一块,结合到之前的terasoluna框架,觉得书本上讲的还是简单阿。我就把我从书本上学到的再结合实际的项目以及网上看到的一些内容,对声明式事务管理做个整理吧。我看得Spring in Action第二版中只提到了用TransactionProxyFactoryBean和<tx:advice/>,定义注释驱动这三种,我承认后两种的内容很好,很强大。但是实际的项目当中
- java 动态代理简单实现
antlove
javahandlerproxydynamicservice
dynamicproxy.service.HelloService
package dynamicproxy.service;
public interface HelloService {
public void sayHello();
}
dynamicproxy.service.impl.HelloServiceImpl
package dynamicp
- JDBC连接数据库
百合不是茶
JDBC编程JAVA操作oracle数据库
如果我们要想连接oracle公司的数据库,就要首先下载oralce公司的驱动程序,将这个驱动程序的jar包导入到我们工程中;
JDBC链接数据库的代码和固定写法;
1,加载oracle数据库的驱动;
&nb
- 单例模式中的多线程分析
bijian1013
javathread多线程java多线程
谈到单例模式,我们立马会想到饿汉式和懒汉式加载,所谓饿汉式就是在创建类时就创建好了实例,懒汉式在获取实例时才去创建实例,即延迟加载。
饿汉式:
package com.bijian.study;
public class Singleton {
private Singleton() {
}
// 注意这是private 只供内部调用
private static
- javascript读取和修改原型特别需要注意原型的读写不具有对等性
bijian1013
JavaScriptprototype
对于从原型对象继承而来的成员,其读和写具有内在的不对等性。比如有一个对象A,假设它的原型对象是B,B的原型对象是null。如果我们需要读取A对象的name属性值,那么JS会优先在A中查找,如果找到了name属性那么就返回;如果A中没有name属性,那么就到原型B中查找name,如果找到了就返回;如果原型B中也没有
- 【持久化框架MyBatis3六】MyBatis3集成第三方DataSource
bit1129
dataSource
MyBatis内置了数据源的支持,如:
<environments default="development">
<environment id="development">
<transactionManager type="JDBC" />
<data
- 我程序中用到的urldecode和base64decode,MD5
bitcarter
cMD5base64decodeurldecode
这里是base64decode和urldecode,Md5在附件中。因为我是在后台所以需要解码:
string Base64Decode(const char* Data,int DataByte,int& OutByte)
{
//解码表
const char DecodeTable[] =
{
0, 0, 0, 0, 0, 0
- 腾讯资深运维专家周小军:QQ与微信架构的惊天秘密
ronin47
社交领域一直是互联网创业的大热门,从PC到移动端,从OICQ、MSN到QQ。到了移动互联网时代,社交领域应用开始彻底爆发,直奔黄金期。腾讯在过去几年里,社交平台更是火到爆,QQ和微信坐拥几亿的粉丝,QQ空间和朋友圈各种刷屏,写心得,晒照片,秀视频,那么谁来为企鹅保驾护航呢?支撑QQ和微信海量数据背后的架构又有哪些惊天内幕呢?本期大讲堂的内容来自今年2月份ChinaUnix对腾讯社交网络运营服务中心
- java-69-旋转数组的最小元素。把一个数组最开始的若干个元素搬到数组的末尾,我们称之为数组的旋转。输入一个排好序的数组的一个旋转,输出旋转数组的最小元素
bylijinnan
java
public class MinOfShiftedArray {
/**
* Q69 旋转数组的最小元素
* 把一个数组最开始的若干个元素搬到数组的末尾,我们称之为数组的旋转。输入一个排好序的数组的一个旋转,输出旋转数组的最小元素。
* 例如数组{3, 4, 5, 1, 2}为{1, 2, 3, 4, 5}的一个旋转,该数组的最小值为1。
*/
publ
- 看博客,应该是有方向的
Cb123456
反省看博客
看博客,应该是有方向的:
我现在就复习以前的,在补补以前不会的,现在还不会的,同时完善完善项目,也看看别人的博客.
我刚突然想到的:
1.应该看计算机组成原理,数据结构,一些算法,还有关于android,java的。
2.对于我,也快大四了,看一些职业规划的,以及一些学习的经验,看看别人的工作总结的.
为什么要写
- [开源与商业]做开源项目的人生活上一定要朴素,尽量减少对官方和商业体系的依赖
comsci
开源项目
为什么这样说呢? 因为科学和技术的发展有时候需要一个平缓和长期的积累过程,但是行政和商业体系本身充满各种不稳定性和不确定性,如果你希望长期从事某个科研项目,但是却又必须依赖于某种行政和商业体系,那其中的过程必定充满各种风险。。。
所以,为避免这种不确定性风险,我
- 一个 sql优化 ([精华] 一个查询优化的分析调整全过程!很值得一看 )
cwqcwqmax9
sql
见 http://www.itpub.net/forum.php?mod=viewthread&tid=239011
Web翻页优化实例
提交时间: 2004-6-18 15:37:49 回复 发消息
环境:
Linux ve
- Hibernat and Ibatis
dashuaifu
Hibernateibatis
Hibernate VS iBATIS 简介 Hibernate 是当前最流行的O/R mapping框架,当前版本是3.05。它出身于sf.net,现在已经成为Jboss的一部分了 iBATIS 是另外一种优秀的O/R mapping框架,当前版本是2.0。目前属于apache的一个子项目了。 相对Hibernate“O/R”而言,iBATIS 是一种“Sql Mappi
- 备份MYSQL脚本
dcj3sjt126com
mysql
#!/bin/sh
# this shell to backup mysql
#
[email protected] (QQ:1413161683 DuChengJiu)
_dbDir=/var/lib/mysql/
_today=`date +%w`
_bakDir=/usr/backup/$_today
[ ! -d $_bakDir ] && mkdir -p
- iOS第三方开源库的吐槽和备忘
dcj3sjt126com
ios
转自
ibireme的博客 做iOS开发总会接触到一些第三方库,这里整理一下,做一些吐槽。 目前比较活跃的社区仍旧是Github,除此以外也有一些不错的库散落在Google Code、SourceForge等地方。由于Github社区太过主流,这里主要介绍一下Github里面流行的iOS库。 首先整理了一份
Github上排名靠
- html wlwmanifest.xml
eoems
htmlxml
所谓优化wp_head()就是把从wp_head中移除不需要元素,同时也可以加快速度。
步骤:
加入到function.php
remove_action('wp_head', 'wp_generator');
//wp-generator移除wordpress的版本号,本身blog的版本号没什么意义,但是如果让恶意玩家看到,可能会用官网公布的漏洞攻击blog
remov
- 浅谈Java定时器发展
hacksin
java并发timer定时器
java在jdk1.3中推出了定时器类Timer,而后在jdk1.5后由Dou Lea从新开发出了支持多线程的ScheduleThreadPoolExecutor,从后者的表现来看,可以考虑完全替代Timer了。
Timer与ScheduleThreadPoolExecutor对比:
1.
Timer始于jdk1.3,其原理是利用一个TimerTask数组当作队列
- 移动端页面侧边导航滑入效果
ini
jqueryWebhtml5cssjavascirpt
效果体验:http://hovertree.com/texiao/mobile/2.htm可以使用移动设备浏览器查看效果。效果使用到jquery-2.1.4.min.js,该版本的jQuery库是用于支持HTML5的浏览器上,不再兼容IE8以前的浏览器,现在移动端浏览器一般都支持HTML5,所以使用该jQuery没问题。HTML文件代码:
<!DOCTYPE html>
<h
- AspectJ+Javasist记录日志
kane_xie
aspectjjavasist
在项目中碰到这样一个需求,对一个服务类的每一个方法,在方法开始和结束的时候分别记录一条日志,内容包括方法名,参数名+参数值以及方法执行的时间。
@Override
public String get(String key) {
// long start = System.currentTimeMillis();
// System.out.println("Be
- redis学习笔记
MJC410621
redisNoSQL
1)nosql数据库主要由以下特点:非关系型的、分布式的、开源的、水平可扩展的。
1,处理超大量的数据
2,运行在便宜的PC服务器集群上,
3,击碎了性能瓶颈。
1)对数据高并发读写。
2)对海量数据的高效率存储和访问。
3)对数据的高扩展性和高可用性。
redis支持的类型:
Sring 类型
set name lijie
get name lijie
set na
- 使用redis实现分布式锁
qifeifei
在多节点的系统中,如何实现分布式锁机制,其中用redis来实现是很好的方法之一,我们先来看一下jedis包中,有个类名BinaryJedis,它有个方法如下:
public Long setnx(final byte[] key, final byte[] value) {
checkIsInMulti();
client.setnx(key, value);
ret
- BI并非万能,中层业务管理报表要另辟蹊径
张老师的菜
大数据BI商业智能信息化
BI是商业智能的缩写,是可以帮助企业做出明智的业务经营决策的工具,其数据来源于各个业务系统,如ERP、CRM、SCM、进销存、HER、OA等。
BI系统不同于传统的管理信息系统,他号称是一个整体应用的解决方案,是融入管理思想的强大系统:有着系统整体的设计思想,支持对所有
- 安装rvm后出现rvm not a function 或者ruby -v后提示没安装ruby的问题
wudixiaotie
function
1.在~/.bashrc最后加入
[[ -s "$HOME/.rvm/scripts/rvm" ]] && source "$HOME/.rvm/scripts/rvm"
2.重新启动terminal输入:
rvm use ruby-2.2.1 --default
把当前安装的ruby版本设为默
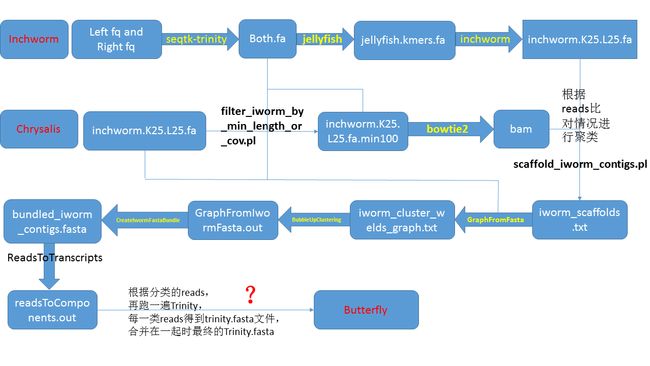 Trinity运行过程.png
Trinity运行过程.png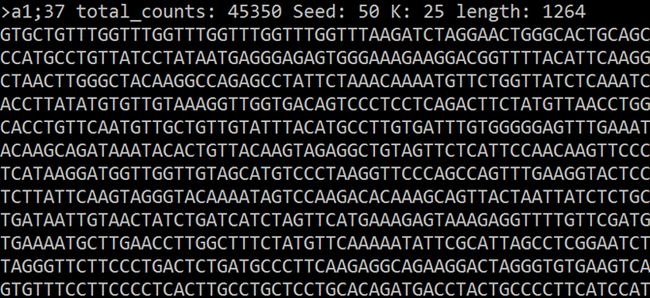 图片.png
图片.png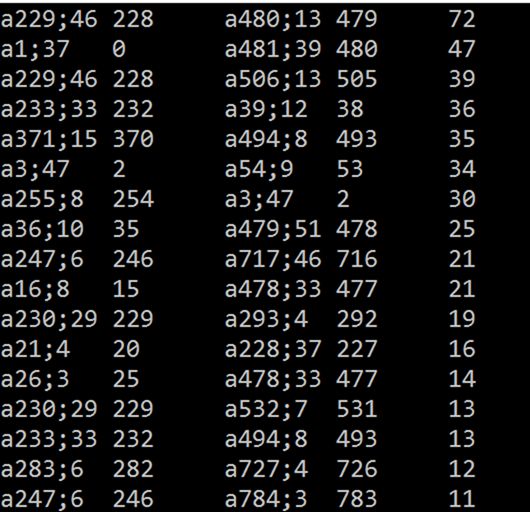 图片.png
图片.png