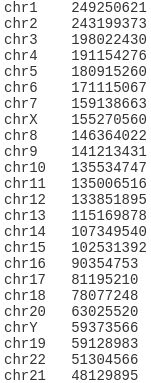
- 基于NXP+FPGA轨道交通3U机箱结构远程输入/输出模块(RIOM)
深圳信迈主板定制专家
轨道交通NXP+FPGAfpga开发人工智能大数据边缘计算运维
基于NXP+FPGA轨道交通6U机箱结构远程输入/输出模块(RIOM)RIOM使得数据通过就近的I/O源输入和输出。也可以直接将I/O源连接到列车计算机(如VCU),可以减少电缆用量从而节约成本。关键特性支持模拟和数字输入/输出。可配置的模块包括DI、DIO、MDO、RDO、AIO、PTI等。接口选项MVBRIOM设备支持MVB/CAN/串行链路三种接口;TRDPRIOM设备知此恨TRDP/CAN
- 【开源代码解读】AI检索系统R1-Searcher通过强化学习RL激励大模型LLM的搜索能力
accurater
人工智能深度学习R1-Searcher
关于R1-Searcher的报告:第一章:引言-AI检索系统的技术演进与R1-Searcher的创新定位1.1信息检索技术的范式转移在数字化时代爆发式增长的数据洪流中,信息检索系统正经历从传统关键词匹配到语义理解驱动的根本性变革。根据IDC的统计,2023年全球数据总量已突破120ZB,其中非结构化数据占比超过80%。这种数据形态的转变对检索系统提出了三个核心的挑战:语义歧义消除:如何准确理解"A
- webgl threejs 云渲染(服务器渲染、后端渲染)解决方案
allenjiao
Threejswebglthreejs云渲染后端渲染服务器渲染云流化三维云渲染
云渲染和流式传输共享三维模型场景1、本地无需高端GPU设备即可提供三维项目渲染云渲染和云流化媒体都可以让3D模型共享变得简单便捷。配备强大GPU的远程服务器早就可以处理密集的处理工作,而专有应用程序,用户也可以从任何个人设备查看全保真模型并与之交互。2、云流媒体实现多终端联动共享价值更高在项目应用场景中,在大屏、电脑、平板、手机和其它移动终端,可以实现多屏联动、远程协助,三维云流化让客户访问时可以
- 【Springboot知识】开发属于自己的中间件健康监测HealthIndicate
问道飞鱼
微服务相关技术springboot中间件后端HealthIndicate
文章目录**一、技术栈****二、项目结构****三、依赖配置(pom.xml)****四、配置文件(application.yml)****五、自定义健康检查实现****1.Redis健康检查****2.Elasticsearch健康检查****3.Kafka健康检查****4.MySQL健康检查****六、自定义健康检查接口(可选)****七、测试与验证****八、高级功能扩展****九、部署
- springboot新手入门搭建项目
stayhungerstayflush
springboot后端java
SpringBoot新手入门指南:从原理到实践一、SpringBoot简介SpringBoot是基于Spring框架的快速开发脚手架,通过约定优于配置的设计理念,简化了Spring应用的初始化搭建和开发过程。主要优势包括:内嵌Web服务器(Tomcat/Jetty)自动配置Spring和第三方库提供生产级监控端点无需XML配置二、核心概念解析1.自动配置(Auto-Configuration)@S
- 开源应用驱动企业新质生产力:Websoft9以EPP+AI+知识库助您领跑未来
开源
开源应用驱动企业新质生产力:Websoft9以EPP+AI+知识库助您领跑未来在数字化转型加速的今天,企业新质生产力的核心已从传统资源投入转向技术驱动的效率革命。开源应用凭借其灵活性、成本优势和技术创新力,成为企业实现这一目标的关键引擎。作为开源技术与行业场景化落地的领航者,Websoft9通过企业应用平台(EPP)、AI智能引擎与知识库系统三位一体的解决方案,助力企业快速构建新一代生产力工具,实
- Web三要素:HTML之ARIA可访问性(3)
双囍菜菜
前端随记前端html服务器ARIA
ARIA:为Web构建数字盲道的技术革命文章目录ARIA:为Web构建数字盲道的技术革命一、屏幕背后的黑暗世界:一个被忽视的用户群体1.1触目惊心的现实案例1.2法律合规的达摩克利斯之剑二、ARIA技术体系的三重维度2.1角色(Roles):定义元素身份常用角色分类2.2属性(Properties):描述元素特征关键属性矩阵2.3状态(States):反映动态变化状态同步机制三、ARIA实战:构建
- 高并发系统的艺术:如何在流量洪峰中游刃有余
架构
作者:京东物流赵勇萍前言我们常说的三高,高并发、高可用、高性能,这些技术是构建现代互联网应用程序所必需的。对于京东618备战来说,所有的中台系统服务,无疑都是围绕着三高来展开的。而对于京东庞大的客户群体,高并发的要求尤为重要。用户对在线服务的需求和期望不断提高,系统的并发处理能力成为衡量其性能和用户体验的关键指标之一。高并发系统不仅仅是大型互联网企业的专利,对于任何希望在市场中占据一席之地的公司来
- Android第二次面试总结(项目拷打实战)
每次的天空
android
MVVM+Jetpack组件落地采用ViewModel+LiveData实现数据驱动开发,将UI逻辑与业务逻辑解耦,通过LiveData的生命周期感知能力避免内存泄漏。使用WorkManager替代传统Service处理后台任务(如数据同步),结合Room数据库实现任务持久化,确保应用被杀后仍能恢复任务。性能优化实战集成Glide加载国风插画,结合自定义三级缓存策略(内存LRU+磁盘缓存+本地资源
- 【JVM】性能监控与调优概述篇
白晨并不是很能熬夜
JVMjvm后端面试java经验分享求职招聘
大家好,我是白晨,一个不是很能熬夜,但是也想日更的人✈。如果喜欢这篇文章,点个赞,关注一下白晨吧!你的支持就是我最大的动力!文章目录JVM性能监控与调优概述篇背景说明生产环境中的问题为什么要调优不同阶段的考虑调优概述监控的依据调优的大方向性能优化的步骤第一步(发现问题):性能监控第二步(排查问题):性能分析第三步(解决问题):性能调优性能评价/测试指标停顿时间(或响应时间)吞吐量并发数内存站用相互
- 运维系列(亲测有效):Docker pull拉取镜像报错“Error response from daemon: Get “https://registry-1.docker.io/v2”解决办法
坦笑&&life
运维运维docker容器
Dockerpull拉取镜像报错“Errorresponsefromdaemon:Get“https://registry-1.docker.io/v2”解决办法一、报错信息二、检查daemon.json文件1.编辑daemon.json2.重启服务三、查看dns解析四、添加host解析五、重新拉取镜像一、报错信息[root@node~]#dockerpullo2oa/o2serverUsingd
- android 新闻客户端和springboot后台开发-网络接口封装(三)
mmsx
android作业源码分享androidspringboot
一、前言android新闻客户端和springboot后台开发(一)-CSDN博客android新闻客户端和springboot后台开发(二)-CSDN博客这篇接前面,写android客户端接口这样方面的实现。okhttp简易封装,方便使用。二、例如注册接口示例UsermUser=newUser(account,password,UserTypeEnum.User.getDesc());Okhtt
- 基于大模型的单纯性孔源性视网膜脱离预测及治疗方案研究报告
LCG元
围术期危险因子预测模型研究人工智能
目录一、引言1.1研究背景与目的1.2国内外研究现状1.3研究方法与创新点二、单纯性孔源性视网膜脱离概述2.1发病机制2.2高危因素2.3临床表现与诊断方法三、大模型在术前预测中的应用3.1模型选择与数据收集3.2术前风险预测指标3.3预测结果分析与验证四、基于预测结果的手术方案制定4.1手术原则与目标4.2不同预测结果下的手术方式选择4.3手术案例分析五、麻醉方案的确定5.1麻醉方式的选择依据5
- C语言编译与链接详解
夜晟洛
c语言开发语言
C语言是一种强大且广泛使用的编程语言。理解其编译和链接过程对于编写高效和可靠的代码至关重要。本文将详细探讨C语言的编译和链接过程,帮助你更好地理解代码从源文件到可执行文件的转变过程。目录一、编译过程概述1.预处理2.编译3.汇编4.链接二、编译与链接示例三、常见问题与最佳实践1.头文件保护2.模块化编程3.静态库和动态库静态库动态库四、总结一、编译过程概述编译过程将C语言源代码转换为机器码,可以分
- 第13章贪心算法
厨 神
贪心算法算法
贪心算法局部最优求得总体最优适用于桌上有6张纸币,面额为10010050505010,问怎么能拿走3张纸币,总面额最大?—拿单位价值最高的只关注局部最优----关注拿一张的最大值拆解-----拿三次最大的纸币不适用于桌面三件物品,每个物品都有重量和价值,wv695733承重为8,求不超过背包承重情况下最大价值只能选一件,能不能得到最大值----选69还剩下二,能选第二件吗?不能选所以不适用,因为不
- Python 科学计算与机器学习入门:NumPy + Scikit-Learn 实战指南
吴师兄大模型
pythonnumpyscikit-learn人工智能开发语言机器学习编程
Langchain系列文章目录01-玩转LangChain:从模型调用到Prompt模板与输出解析的完整指南02-玩转LangChainMemory模块:四种记忆类型详解及应用场景全覆盖03-全面掌握LangChain:从核心链条构建到动态任务分配的实战指南04-玩转LangChain:从文档加载到高效问答系统构建的全程实战05-玩转LangChain:深度评估问答系统的三种高效方法(示例生成、手
- C语言:整数、浮点数在内存中的存储
代码AC不AC
学习分享c语言
hello,我又来了!~内存存储1、整数在内存中的存储2、浮点数在内存中的存储3、2中的例题解释1、整数在内存中的存储我们知道:整数的表达式有三种,即:原码、反码和补码。正整数的原码、反码和补码都相同。负整数的三种表达式各不相同。原码:将数值按照负数的形式翻译成二进制得到原码。反码:原码的符号位(首位)不变,其他位依次按位取反就得到反码。补码:反码+1。对整型来说,数据存放的是二进制的补码。原因:
- 微信小程序根据不同用户切换不同`TabBar`,简单易懂
Duaigi
小程序小程序
现有需求:小程序用户有三种身份(公众、运维人员、领导),根据不同用户身份显示不同的tabbar众所周知微信小程序全局文件app.json里面的"tabBar"里面的list只能放置2-5个,要想实现3个tabbar,必须得复用tabbar,三种身份都需要个人中心,剩下的是长列表(两个),表单,图表刚好是5个,废话少说,上代码代码有点长,建议仔细看一下1全局.app.jsontabbar里面的sus
- 数据仓库有哪些建模方法?
BenBen尔
#数据仓库数据仓库大数据
数据仓库的建模方法主要分为关系建模和多维建模两大类,不同方法适用于不同的业务场景和目标。以下是常见的建模方法及其特点:一、关系建模(规范化建模)基于关系型数据库的规范化理论,强调减少数据冗余,适合复杂的企业级数据仓库(EDW)。第三范式(3NF)定义:通过规范化将数据分解为多个关联表,确保每个字段仅依赖主键。优点:数据冗余低,一致性高,适合复杂事务处理。缺点:查询需要多表关联,性能较低;业务理解成
- python start函数_Python中10个常用的内置函数
半残大叔霁天
pythonstart函数
大家好,我是小张在3.8版本中,Python解释器有近69个内置函数可供使用,有了它们能极大地提高编码效率,数量虽然不少,但在日常搬砖中只用到其中一部分,根据使用频率和用法,这里列出来几个本人认为不错的内置函数,结合一些例子介绍给大家complex()返回一个形如a+bj的复数,传入参数分为三种情况:参数为空时,返回0j参数为字符串时,将字符串表达式解释为复数形式并返回参数为两个整数(a,b)时,
- 从前端视角理解消息队列:核心问题与实战指南
秋水为渡
前端
消息队列(MessageQueue)是现代分布式系统的核心组件之一,它在前后端协作、系统解耦、流量削峰等场景中发挥着重要作用。本文从前端开发者视角出发,解析消息队列的关键问题,并结合实际场景给出解决方案。一、为什么要使用消息队列?1.前端常见场景异步任务处理:用户行为日志上报、实时通知推送流量削峰:应对秒杀活动、大文件上传等瞬时高并发场景系统解耦:前端与后端服务、第三方服务之间的松耦合通信2.前端
- 数据分析大数据面试题大杂烩01
爱学习的菜鸟罢了
大数据flink大数据面试hivehadoopkafka
互联网:通过埋点实时计算用户浏览频次用优惠券等措施吸引用户,通过历史信息用非智能学习的title方式构造用户画像(抖音,京东)电信,银行统计营收和针对用户的个人画像:处理大量非实时数据政府:健康码,扫码之后确诊,找出与确诊对象有关联的人订单订单表(除商品以外所有信息),商品详情表,通过搜集用户title进行定制化推荐点击流数据通过埋点进行用户点击行为分析FLINK一般用来做实时SPARK一般用来做
- 前端请求全面解析:AJAX、Axios 与 Fetch 的使用详解与代码示例
软件工匠师
前端ajaxjavascript
前端请求全面解析:AJAX、Axios与Fetch的使用详解与代码示例前端请求全面解析:AJAX、Axios与Fetch的使用详解与代码示例1.AJAX——传统的异步请求1.1基本用法示例1.2AJAX特点2.FetchAPI——现代化请求方案2.1基本用法示例2.2Fetch特点3.Axios——第三方HTTP请求库3.1安装Axios3.2基本用法示例3.3Axios特点4.总结前端请求全面解
- 海量数据查询加速:Presto、Trino、Apache Arrow
晴天彩虹雨
apache大数据hive数据仓库
1.引言在大数据分析场景下,查询速度往往是影响业务决策效率的关键因素。随着数据量的增长,传统的行存储数据库难以满足低延迟的查询需求,因此,基于列式存储、向量化计算等技术的查询引擎应运而生。本篇文章将深入探讨Presto、Trino、ApacheArrow三种主流的查询优化工具,剖析其核心机制,并通过案例分析展示它们在实际业务中的应用。2.Presto:分布式SQL查询引擎2.1Presto介绍Pr
- 二叉树的所有路径(leetcode 257
JohnFF
leetcodelinux算法
leetcode系列文章目录一、核心操作二、外层配合操作三、核心模式代码总结使用递归法一、核心操作1.判断是不是叶子节点(该节点的左右子节点都为空2.收获该路径(将储存的节点一个一个拿出来,用->连接if(cur->left==nullptr&&cur->right==nullptr){stringspath;for(inti=0;i";}spath+=to_string(path[path.si
- 合并二叉树 迭代(leetcode 617
JohnFF
leetcode算法职场和发展
leetcode系列文章目录一、核心操作二、外层配合操作三、核心模式代码总结一、核心操作1.将右树的值加到左树上2.对两棵树的子节点进行筛选,如果都有则都加进去,如果左树没有则将右数的节点指针赋给左树,如果左树有右树没有则不用管提示:小白个人理解,如有错误敬请谅解!二、外层配合操作1.确保root1和root2都有值,所以当一棵树为空则返回另外一棵树三、核心模式代码代码如下:classSoluti
- 数组总和 (leetcode 40
JohnFF
leetcode算法职场和发展
leetcode系列文章目录一、核心操作二、外层配合操作三、核心模式代码总结去重方式和之前三数之和一样,也可以用used数组去重,但本次尝试使用set去重一、核心操作如果count为0了,则证明正好减到了0,就可以收获,并返回建立unordered_set开始循环,如果在set中能够搜寻到当前的数字,说明已经重复了,则直接进行下一次的循环,如果没有找到,则说明这是一个没有重复的新数字,将其加入se
- Vue动态组件完全指南:原理、使用场景与最佳实践
北辰alk
前端vuevue.jsjavascript前端
文章目录一、什么是动态组件?核心特性:二、基本使用方式1.基础语法2.组件注册方式3.动态组件生命周期三、六大典型应用场景1.标签页切换系统2.多步骤表单流程3.动态仪表盘4.权限驱动视图5.插件系统集成6.服务端驱动界面四、高级使用技巧1.状态保持方案2.动态Props传递3.异步组件加载4.过渡动画支持五、性能优化策略1.缓存策略对比2.代码分割配置3.内存管理示例六、常见问题解决方案1.组件
- Spring Cache的基本使用
奇怪的大象
面试学习路线阿里巴巴springjava后端
文章目录一、概述二、SpringCache的使用2.1环境搭建2.2缓存的读模式@Cacheable2.3自定义缓存配置
[email protected]@CacheEvict删除缓存2.6@Caching多个操作三、SpringCache的不足一、概述常见的缓存的框架有Redis、Memcached、Guava、Caffeine等等,各有各的优势。如果我们的程序想要使用缓存,就要与这些框架耦合。聪明
- 【考研计算机网络】课堂笔记4 第四章 网络层_Network Layer
刘鑫磊up
#操作系统计算机网络计算机网络
文章目录:一:网络层的功能1.异构网络互联2.路由与转发功能3.拥塞控制二:数据交换方式三:路由算法1.静态路由与动态路由1.1静态路由算法(又称非自适应路由算法)1.2动态路由算法(又称自适应路由算法)2.动态路由算法2.1距离-向量路由算法2.2链路状态路由算法2.3层次路由四:IPV41.概述2.IPV4分组2.1IPV4分组格式2.2IP数据报分片2.3网络层转发分组的流程3IPV4地址与
- [星球大战]阿纳金的背叛
comsci
本来杰迪圣殿的长老是不同意让阿纳金接受训练的.........
但是由于政治原因,长老会妥协了...这给邪恶的力量带来了机会
所以......现代的地球联邦接受了这个教训...绝对不让某些年轻人进入学院
- 看懂它,你就可以任性的玩耍了!
aijuans
JavaScript
javascript作为前端开发的标配技能,如果不掌握好它的三大特点:1.原型 2.作用域 3. 闭包 ,又怎么可以说你学好了这门语言呢?如果标配的技能都没有撑握好,怎么可以任性的玩耍呢?怎么验证自己学好了以上三个基本点呢,我找到一段不错的代码,稍加改动,如果能够读懂它,那么你就可以任性了。
function jClass(b
- Java常用工具包 Jodd
Kai_Ge
javajodd
Jodd 是一个开源的 Java 工具集, 包含一些实用的工具类和小型框架。简单,却很强大! 写道 Jodd = Tools + IoC + MVC + DB + AOP + TX + JSON + HTML < 1.5 Mb
Jodd 被分成众多模块,按需选择,其中
工具类模块有:
jodd-core &nb
- SpringMvc下载
120153216
springMVC
@RequestMapping(value = WebUrlConstant.DOWNLOAD)
public void download(HttpServletRequest request,HttpServletResponse response,String fileName) {
OutputStream os = null;
InputStream is = null;
- Python 标准异常总结
2002wmj
python
Python标准异常总结
AssertionError 断言语句(assert)失败 AttributeError 尝试访问未知的对象属性 EOFError 用户输入文件末尾标志EOF(Ctrl+d) FloatingPointError 浮点计算错误 GeneratorExit generator.close()方法被调用的时候 ImportError 导入模块失
- SQL函数返回临时表结构的数据用于查询
357029540
SQL Server
这两天在做一个查询的SQL,这个SQL的一个条件是通过游标实现另外两张表查询出一个多条数据,这些数据都是INT类型,然后用IN条件进行查询,并且查询这两张表需要通过外部传入参数才能查询出所需数据,于是想到了用SQL函数返回值,并且也这样做了,由于是返回多条数据,所以把查询出来的INT类型值都拼接为了字符串,这时就遇到问题了,在查询SQL中因为条件是INT值,SQL函数的CAST和CONVERST都
- java 时间格式化 | 比较大小| 时区 个人笔记
7454103
javaeclipsetomcatcMyEclipse
个人总结! 不当之处多多包含!
引用 1.0 如何设置 tomcat 的时区:
位置:(catalina.bat---JAVA_OPTS 下面加上)
set JAVA_OPT
- 时间获取Clander的用法
adminjun
Clander时间
/**
* 得到几天前的时间
* @param d
* @param day
* @return
*/
public static Date getDateBefore(Date d,int day){
Calend
- JVM初探与设置
aijuans
java
JVM是Java Virtual Machine(Java虚拟机)的缩写,JVM是一种用于计算设备的规范,它是一个虚构出来的计算机,是通过在实际的计算机上仿真模拟各种计算机功能来实现的。Java虚拟机包括一套字节码指令集、一组寄存器、一个栈、一个垃圾回收堆和一个存储方法域。 JVM屏蔽了与具体操作系统平台相关的信息,使Java程序只需生成在Java虚拟机上运行的目标代码(字节码),就可以在多种平台
- SQL中ON和WHERE的区别
avords
SQL中ON和WHERE的区别
数据库在通过连接两张或多张表来返回记录时,都会生成一张中间的临时表,然后再将这张临时表返回给用户。 www.2cto.com 在使用left jion时,on和where条件的区别如下: 1、 on条件是在生成临时表时使用的条件,它不管on中的条件是否为真,都会返回左边表中的记录。
- 说说自信
houxinyou
工作生活
自信的来源分为两种,一种是源于实力,一种源于头脑.实力是一个综合的评定,有自身的能力,能利用的资源等.比如我想去月亮上,要身体素质过硬,还要有飞船等等一系列的东西.这些都属于实力的一部分.而头脑不同,只要你头脑够简单就可以了!同样要上月亮上,你想,我一跳,1米,我多跳几下,跳个几年,应该就到了!什么?你说我会往下掉?你笨呀你!找个东西踩一下不就行了吗?
无论工作还
- WEBLOGIC事务超时设置
bijian1013
weblogicjta事务超时
系统中统计数据,由于调用统计过程,执行时间超过了weblogic设置的时间,提示如下错误:
统计数据出错!
原因:The transaction is no longer active - status: 'Rolling Back. [Reason=weblogic.transaction.internal
- 两年已过去,再看该如何快速融入新团队
bingyingao
java互联网融入架构新团队
偶得的空闲,翻到了两年前的帖子
该如何快速融入一个新团队,有所感触,就记下来,为下一个两年后的今天做参考。
时隔两年半之后的今天,再来看当初的这个博客,别有一番滋味。而我已经于今年三月份离开了当初所在的团队,加入另外的一个项目组,2011年的这篇博客之后的时光,我很好的融入了那个团队,而直到现在和同事们关系都特别好。大家在短短一年半的时间离一起经历了一
- 【Spark七十七】Spark分析Nginx和Apache的access.log
bit1129
apache
Spark分析Nginx和Apache的access.log,第一个问题是要对Nginx和Apache的access.log文件进行按行解析,按行解析就的方法是正则表达式:
Nginx的access.log解析正则表达式
val PATTERN = """([^ ]*) ([^ ]*) ([^ ]*) (\\[.*\\]) (\&q
- Erlang patch
bookjovi
erlang
Totally five patchs committed to erlang otp, just small patchs.
IMO, erlang really is a interesting programming language, I really like its concurrency feature.
but the functional programming style
- log4j日志路径中加入日期
bro_feng
javalog4j
要用log4j使用记录日志,日志路径有每日的日期,文件大小5M新增文件。
实现方式
log4j:
<appender name="serviceLog"
class="org.apache.log4j.RollingFileAppender">
<param name="Encoding" v
- 读《研磨设计模式》-代码笔记-桥接模式
bylijinnan
java设计模式
声明: 本文只为方便我个人查阅和理解,详细的分析以及源代码请移步 原作者的博客http://chjavach.iteye.com/
/**
* 个人觉得关于桥接模式的例子,蜡笔和毛笔这个例子是最贴切的:http://www.cnblogs.com/zhenyulu/articles/67016.html
* 笔和颜色是可分离的,蜡笔把两者耦合在一起了:一支蜡笔只有一种
- windows7下SVN和Eclipse插件安装
chenyu19891124
eclipse插件
今天花了一天时间弄SVN和Eclipse插件的安装,今天弄好了。svn插件和Eclipse整合有两种方式,一种是直接下载插件包,二种是通过Eclipse在线更新。由于之前Eclipse版本和svn插件版本有差别,始终是没装上。最后在网上找到了适合的版本。所用的环境系统:windows7JDK:1.7svn插件包版本:1.8.16Eclipse:3.7.2工具下载地址:Eclipse下在地址:htt
- [转帖]工作流引擎设计思路
comsci
设计模式工作应用服务器workflow企业应用
作为国内的同行,我非常希望在流程设计方面和大家交流,刚发现篇好文(那么好的文章,现在才发现,可惜),关于流程设计的一些原理,个人觉得本文站得高,看得远,比俺的文章有深度,转载如下
=================================================================================
自开博以来不断有朋友来探讨工作流引擎该如何
- Linux 查看内存,CPU及硬盘大小的方法
daizj
linuxcpu内存硬盘大小
一、查看CPU信息的命令
[root@R4 ~]# cat /proc/cpuinfo |grep "model name" && cat /proc/cpuinfo |grep "physical id"
model name : Intel(R) Xeon(R) CPU X5450 @ 3.00GHz
model name :
- linux 踢出在线用户
dongwei_6688
linux
两个步骤:
1.用w命令找到要踢出的用户,比如下面:
[root@localhost ~]# w
18:16:55 up 39 days, 8:27, 3 users, load average: 0.03, 0.03, 0.00
USER TTY FROM LOGIN@ IDLE JCPU PCPU WHAT
- 放手吧,就像不曾拥有过一样
dcj3sjt126com
内容提要:
静悠悠编著的《放手吧就像不曾拥有过一样》集结“全球华语世界最舒缓心灵”的精华故事,触碰生命最深层次的感动,献给全世界亿万读者。《放手吧就像不曾拥有过一样》的作者衷心地祝愿每一位读者都给自己一个重新出发的理由,将那些令你痛苦的、扛起的、背负的,一并都放下吧!把憔悴的面容换做一种清淡的微笑,把沉重的步伐调节成春天五线谱上的音符,让自己踏着轻快的节奏,在人生的海面上悠然漂荡,享受宁静与
- php二进制安全的含义
dcj3sjt126com
PHP
PHP里,有string的概念。
string里,每个字符的大小为byte(与PHP相比,Java的每个字符为Character,是UTF8字符,C语言的每个字符可以在编译时选择)。
byte里,有ASCII代码的字符,例如ABC,123,abc,也有一些特殊字符,例如回车,退格之类的。
特殊字符很多是不能显示的。或者说,他们的显示方式没有标准,例如编码65到哪儿都是字母A,编码97到哪儿都是字符
- Linux下禁用T440s,X240的一体化触摸板(touchpad)
gashero
linuxThinkPad触摸板
自打1月买了Thinkpad T440s就一直很火大,其中最让人恼火的莫过于触摸板。
Thinkpad的经典就包括用了小红点(TrackPoint)。但是小红点只能定位,还是需要鼠标的左右键的。但是自打T440s等开始启用了一体化触摸板,不再有实体的按键了。问题是要是好用也行。
实际使用中,触摸板一堆问题,比如定位有抖动,以及按键时会有飘逸。这就导致了单击经常就
- graph_dfs
hcx2013
Graph
package edu.xidian.graph;
class MyStack {
private final int SIZE = 20;
private int[] st;
private int top;
public MyStack() {
st = new int[SIZE];
top = -1;
}
public void push(i
- Spring4.1新特性——Spring核心部分及其他
jinnianshilongnian
spring 4.1
目录
Spring4.1新特性——综述
Spring4.1新特性——Spring核心部分及其他
Spring4.1新特性——Spring缓存框架增强
Spring4.1新特性——异步调用和事件机制的异常处理
Spring4.1新特性——数据库集成测试脚本初始化
Spring4.1新特性——Spring MVC增强
Spring4.1新特性——页面自动化测试框架Spring MVC T
- 配置HiveServer2的安全策略之自定义用户名密码验证
liyonghui160com
具体从网上看
http://doc.mapr.com/display/MapR/Using+HiveServer2#UsingHiveServer2-ConfiguringCustomAuthentication
LDAP Authentication using OpenLDAP
Setting
- 一位30多的程序员生涯经验总结
pda158
编程工作生活咨询
1.客户在接触到产品之后,才会真正明白自己的需求。
这是我在我的第一份工作上面学来的。只有当我们给客户展示产品的时候,他们才会意识到哪些是必须的。给出一个功能性原型设计远远比一张长长的文字表格要好。 2.只要有充足的时间,所有安全防御系统都将失败。
安全防御现如今是全世界都在关注的大课题、大挑战。我们必须时时刻刻积极完善它,因为黑客只要有一次成功,就可以彻底打败你。 3.
- 分布式web服务架构的演变
自由的奴隶
linuxWeb应用服务器互联网
最开始,由于某些想法,于是在互联网上搭建了一个网站,这个时候甚至有可能主机都是租借的,但由于这篇文章我们只关注架构的演变历程,因此就假设这个时候已经是托管了一台主机,并且有一定的带宽了,这个时候由于网站具备了一定的特色,吸引了部分人访问,逐渐你发现系统的压力越来越高,响应速度越来越慢,而这个时候比较明显的是数据库和应用互相影响,应用出问题了,数据库也很容易出现问题,而数据库出问题的时候,应用也容易
- 初探Druid连接池之二——慢SQL日志记录
xingsan_zhang
日志连接池druid慢SQL
由于工作原因,这里先不说连接数据库部分的配置,后面会补上,直接进入慢SQL日志记录。
1.applicationContext.xml中增加如下配置:
<bean abstract="true" id="mysql_database" class="com.alibaba.druid.pool.DruidDataSourc