这篇文章来练习一下ATAC-seq分析。ATAC-seq和CHIP-seq的分析非常相似,CHIP-seq检测的是抗体结合的DNA序列;而ATAC-seq检测的是“易接近”的DNA序列。ATAC-seq原理:ATAC-seq的原理及应用。
数据来源文献:
TGF-β-Induced Quiescence Mediates Chemoresistance of Tumor-Propagating Cells in Squamous Cell Carcinoma. Cell Stem Cell 2017 Nov 2;21(5):650-664.e8.
数据链接:here
(一)数据下载
这里我是直接下载的fastq文件,fastq文件可以到EBI网站下载,根据之前的实战练习,批量下载的方法请见:单细胞测序实战(第一部分)。
按照之前的方法,要先拿到下载的链接list(这篇文献的ATAC-seq测序是双端测序,所以每一个样品是两个fastq文件):

$ sed -e 's/ftp.sra.ebi.ac.uk//g' list.txt >> list_2.txt
$ head list_2.txt
/vol1/fastq/SRR407/004/SRR4071854/SRR4071854_1.fastq.gz
/vol1/fastq/SRR407/005/SRR4071855/SRR4071855_1.fastq.gz
/vol1/fastq/SRR407/006/SRR4071856/SRR4071856_1.fastq.gz
/vol1/fastq/SRR407/007/SRR4071857/SRR4071857_1.fastq.gz
/vol1/fastq/SRR407/008/SRR4071858/SRR4071858_1.fastq.gz
/vol1/fastq/SRR407/009/SRR4071859/SRR4071859_1.fastq.gz
/vol1/fastq/SRR407/000/SRR4071860/SRR4071860_1.fastq.gz
/vol1/fastq/SRR407/001/SRR4071861/SRR4071861_1.fastq.gz
/vol1/fastq/SRR407/004/SRR4071854/SRR4071854_2.fastq.gz
/vol1/fastq/SRR407/005/SRR4071855/SRR4071855_2.fastq.gz
然后利用ascp批量下载(批量下载的代码以及用法见:单细胞测序实战(第一部分)):
$ ascp -QT -l 300m -P33001 -k1 -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh --mode recv --host fasp.sra.ebi.ac.uk --user era-fasp --file-list list_2.txt /media/yanfang/FYWD/ATAC/ATAC_fastq/
SRR4071854_1.fastq.gz 100% 764MB 22.6Mb/s 05:19
SRR4071855_1.fastq.gz 100% 848MB 26.3Mb/s 10:49
SRR4071856_1.fastq.gz 100% 817MB 21.9Mb/s 16:08
SRR4071857_1.fastq.gz 100% 733MB 22.2Mb/s 21:06
SRR4071858_1.fastq.gz 100% 860MB 14.3Mb/s 26:49
SRR4071859_1.fastq.gz 100% 921MB 22.0Mb/s 33:15
SRR4071860_1.fastq.gz 100% 704MB 22.1Mb/s 38:20
SRR4071861_1.fastq.gz 100% 905MB 12.5Mb/s 45:10
SRR4071854_2.fastq.gz 100% 784MB 21.6Mb/s 51:17
SRR4071855_2.fastq.gz 100% 878MB 21.7Mb/s 57:54
SRR4071856_2.fastq.gz 100% 852MB 24.1Mb/s 1:03:26
SRR4071857_2.fastq.gz 100% 761MB 18.8Mb/s 1:08:17
SRR4071858_2.fastq.gz 100% 892MB 16.2Mb/s 1:13:59
SRR4071859_2.fastq.gz 100% 954MB 20.5Mb/s 1:20:38
SRR4071860_2.fastq.gz 100% 720MB 23.9Mb/s 1:25:55
SRR4071861_2.fastq.gz 100% 942MB 19.9Mb/s 1:32:25
Completed: 13662882K bytes transferred in 5545 seconds
(20181K bits/sec), in 16 files.
(二)fastqc质控
经过fastqc进行质量评估后,发现这些测序结果都比较好,都像下面这样:
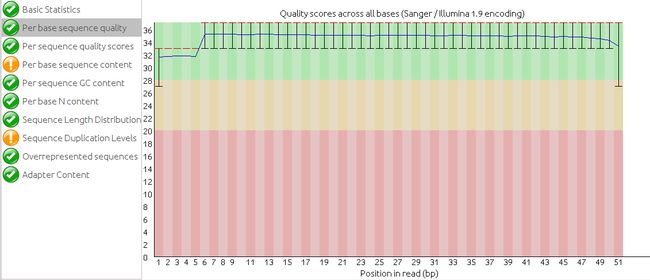
基本上不用trim。所以就直接进行下面的步骤了。
(三)比对
在原文献中,关于ATAC数据的处理是这样描述的:
Mouse assembly version mm10/NCBI m38 build were used for sequence alignments with bowtie (https://www.ncbi.nlm.nih.gov/pubmed/19261174) version 2.1.0. Sequences with MAPQ scores < 30 were removed with samtools (https://www.ncbi.nlm.nih.gov/pubmed/19505943) and duplicates were removed with picard-tools version 1.88 (http://broadinstitute.github.io/picard).
原文作者用了bowtie做的比对,基因组是mm10,比对的结果用MAPQ值过滤了一下,小于30的就都过滤掉了。duplicates用picard-tools过滤。
首先来说比对的软件,原作者用了bowtie,我电脑里安装的是bowtie2。我查了一下bowtie和bowtie2的区别([转] bowtie和bowtie2使用条件区别及用法):
要说明的是,bowtie2并不是bowtie的升级版,它们是独立的、不同的两个软件。
1.bowtie1出现的早,所以对于测序长度在50bp以下的序列效果不错,而bowtie2主要针对的是长度在50bp以上的测序的。
2.Bowtie 2支持有空位的比对
3.Bowtie 2支持局部比对,也可以全局比对
4.Bowtie 2对最长序列没有要求,但是Bowtie 1最长不能超过1000bp。
5.Bowtie 2 allows alignments to [overlap ambiguous characters] (e.g. Ns) in the reference. Bowtie 1 does not.
6.Bowtie 2不能比对colorspace reads.
这篇文献的测序长度是51bp,感觉用哪个都可以。所以我还是用已经安装的bowtie2吧。
比对的代码:
#举个例子
$ bowtie2 -p 6 -x /media/yanfang/FYWD/ATAC/ref_index_mm10_bt/mm10 -1 /media/yanfang/FYWD/ATAC/ATAC_fastq/SRR4071854_1.fastq.gz -2 /media/yanfang/FYWD/ATAC/ATAC_fastq/SRR4071854_2.fastq.gz -S /media/yanfang/FYWD/ATAC/sam_files/SCC_2_CD71hi_GFPhi.sam
28645146 reads; of these:
28645146 (100.00%) were paired; of these:
7312945 (25.53%) aligned concordantly 0 times
13281633 (46.37%) aligned concordantly exactly 1 time
8050568 (28.10%) aligned concordantly >1 times
----
7312945 pairs aligned concordantly 0 times; of these:
2682841 (36.69%) aligned discordantly 1 time
----
4630104 pairs aligned 0 times concordantly or discordantly; of these:
9260208 mates make up the pairs; of these:
5110594 (55.19%) aligned 0 times
1128254 (12.18%) aligned exactly 1 time
3021360 (32.63%) aligned >1 times
91.08% overall alignment rate
(四)sam文件过滤
然后过滤,根据文献中要求的(保留MAPQ>=30的):
#一共有8个文件,写个小脚本
#!/bin/bash
for i in SCC*
do
samtools view -bSq 30 $i > $i.filtered.bam
done

(五)过滤duplicates
在文献里,作者标明,使用picard-tools(1.88)去除duplicates。
安装picard
picard是GATK里的一个工具,所以我们要安装GATK:
$ wget https://github.com/broadinstitute/gatk/releases/download/4.1.4.1/gatk-4.1.4.1.zip
$ unzip gatk-4.1.4.1.zip #解压
$ ./gatk --help #测试是否安装成功
#弹出一堆使用说明,就代表安装成功了,只需要再添加到环境变量里就行了
$ echo 'export PATH=/media/yanfang/FYWD/ATAC/gatk/gatk-4.1.4.1:$PATH' >> ~/.bashrc
$ source ~/.bashrc
安装好了GATK,我查了一下怎么用这个软件,官网:here
Usage example:
$ java -jar picard.jar MarkDuplicates \
I=input.bam \
O=marked_duplicates.bam \
M=marked_dup_metrics.txt
官网对这款软件的介绍主要如下(不是翻译,只是捡重点来说):
MarkDuplicates (Picard)
主要功能:鉴定duplicate reads (可以用sam文件,也可以用bam文件)
MarkDuplicates输出文件会产生一个新的sam或者bam文件,同时会产生一个txt文件,这个txt文件里包含单端或者双端测序reads里的duplicates。
举个栗子:
$ java -jar /media/yanfang/FYWD/ATAC/gatk/gatk-4.1.4.1/gatk-package-4.1.4.1-local.jar MarkDuplicates \
-I=SCC_1_CD71hi_GFPhi.sam.filtered.bam \
-O=SCC_1_CD71hi_GFPhi.rmdup.bam \
--REMOVE_SEQUENCING_DUPLICATES true \
-M=marked_dup_metrics.txt
#这里要注意的是,在java -jar后面,跟的是你安装gatk的路径,因为我发现即便你修改了环境变量,但还是不能在其他地方调用,你需要指定你安装的路径
然后就是漫长的等待了。。。
然而等了一宿,也没跑完一个样品???可能我电脑内存被我折腾的没多少了,所以我在网上查了一下,有木有其他方法可以提高picard的速度,或者有木有其他软件也可以remove duplicates。
对于前者的答案,网上有写:提高内存(跟没说一样)。。。
而对于后者的答案,有些新发现:
我在网上查了一下,发现有一片文章讲到:picard去duplicates的速度非!常!的!慢!(嫌picard去重复太慢?快来试试这两个高效快速的工具吧~)现在很多人使用另外两种软件进行remove duplicates。一个是samblaster,另一个是sambamba。网上的教程里(给学徒的ATAC-seq数据实战)使用的也是sambamba。在筛选标准不变的前提下速度能提升30倍以上。听着很牛的样子。所以我决定用sambamba来试一下,看看是不是传说中那样快。
sambamba的官网:Sambamba: process your BAM data faster!
安装sambamba:
这个安装和一般的软件不一样,如果你傻乎乎的下载,解压,再添加环境变量,是安不上的。安装过程是这样的(我弄了一个晚上加一个上午,报错报的我头大。参考:https://github.com/biod/sambamba#getting-help):
#首先你需要下载一个依赖软件,没有这个软件的话是没法安装sambamba的,网站https://github.com/ldc-developers/ldc/releases/可以下载最新版
$ wget https://github.com/ldc-developers/ldc/releases/download/v1.19.0-beta2/ldc2-1.19.0-beta2-linux-x86_64.tar.xz
$ tar xvjf ldc2-1.19.0-beta2-linux-x86_64.tar.xz
$ cd ldc2-1.19.0-beta2-linux-x86_64/
$ echo 'export PATH=/media/yanfang/FYWD/ATAC/sambamba/ldc2-1.19.0-beta2-linux-x86_64/bin:$PATH' >> ~/.bashrc
$ source ~/.bashrc
#接下来你就可以安装sambamba了
$ git clone --recursive https://github.com/biod/sambamba.git
$ cd sambamba
$ make
$ echo 'export PATH=/media/yanfang/FYWD/ATAC/sambamba/sambamba/bin:$PATH' >> ~/.bashrc
$ source ~/.bashrc
然后当你觉得可以运行sambamba的时候,你会发现:
$ sambamba -view
sambamba: command not found
有没有很气!明明安装后已经加入环境变量了,为什么还不能运行呢?后来我试了无数遍才发现,你需要加上软件的版本号!!!
#像这样:
$ sambamba-0.7.1 -view
#然后它才会弹出来使用说明
sambamba 0.7.1
by Artem Tarasov and Pjotr Prins (C) 2012-2019
LDC 1.19.0-beta2 / DMD v2.089.0 / LLVM9.0.0 / bootstrap LDC - the LLVM D compiler (1.19.0-beta2)
Usage: sambamba [command] [args...]
Available commands:
view view contents and convert from one format
to another (SAM/BAM/CRAM/JSON/UNPACK)
index build index (BAI)
merge merge files (BAM)
sort sort file (BAM)
slice slice file (BAM using BED)
markdup mark or remove duplicates (BAM)
subsample subsample (BAM)
flagstat output statistics (BAM)
depth output statistics (BAM)
validate simple validator (BAM)
No longer recommended:
mpileup parallel execution of samtools (BAM)
To get help on a particular command, call it without args.
Global options
-q quiet mode (do not show banner)
For bug reports and feature requests see
https://github.com/biod/
知道了用法,下面就开始使用它来remove duplicates了:
#因为有8个sam文件,所以写个小脚本
#!/bin/bash
markdup_files=$(ls /media/yanfang/FYWD/ATAC/sam_files/*.filtered.bam)
for i in ${markdup_files}
do
sambamba-0.7.1 markdup --overflow-list-size 600000 --tmpdir='./' -r $i $i.rmdup.bam #这里r的意思是,不标记duplicates,而是直接去掉duplicates。
sambamba-0.7.1 sort $i.rmdup.bam -o $i.rmdup_sorted.bam
sambamba-0.7.1 index $i.rmdup_sorted.bam
done
##--overflow-list-size=OVERFLOWLISTSIZE: size of the overflow list where reads, thrown away from the hash table, get a second chance to meet their pairs (default is 200000 reads); increasing the size reduces the number of temporary files created
##去掉duplicates后,要sort一下,为了之后构建索引index
速度果然快的惊人~做饭加吃饭的时间就把8个sam文件remove duplicates,还运行完了sort和构建索引。比picard快了不知道多少倍啊!
(六)IGV viewer文件(tdf文件)的生成
igvtools是最常用的NGS数据可视化工具之一,可以展示序列比对,拷贝数变异,突变位点等多种数据的分布。TDF格式采用了滑动窗口的方式,用窗口内的数据的平均值来代表该窗口的结果,最终生成了一个二进制的文件(参考使用igvtools可视化测序深度分布)。
根据文献的方法部分对ATAC-seq的IGV可视化的细节说明:
IGV viewer files were generated with igvtools count -z 5 -w 25 -e 250 (https://www.ncbi.nlm.nih.gov/pubmed/21221095)
IGV的安装我就不解释了,网上一堆安装的详细步骤。这里因为我也是第一次用IGV来生成tdf文件,所以主要是探索一下怎么用IGV生成tdf文件。
首先打开你的IGV:
在最上面的工具栏一行里,找到"Tools"这个选项,点击"runIGVtools":
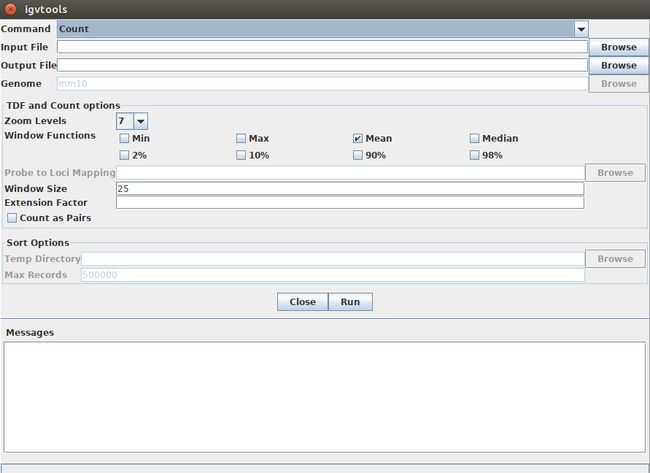
文献里指出他们用的command是count,也就是最上面的一行选项。往下依次看,input file就是你sort好的bam文件(要注意的是,这里虽然不用索引文件,但是你必须有它,不然的话是没法进行下一步分析的)。你输入input file之后,它会在下一行的output file里自动弹出输出文件的路径和文件名(tdf文件),你也可以修改文件名。再下面就是参数的选择了,根据文献说明:zoom levels用5,window size用25,extension factor用250。点击Run,就运行了。一个文件大概不到半分钟的样子。
生成好了tdf文件后,再回到IGV主页面上,工具栏最左面的file里面,选择load from files。把你生成好的tdf文件导入进来,像这样:
随便搜索一个基因,看看附近有没有peak:
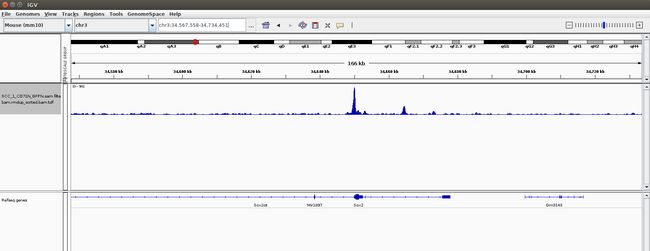
这里说明一下:IGV也支持sam和bam文件,但是为什么不用那两个文件直接导入看peak呢?自己试一下就知道了,电脑配置差一点的就祈祷不要死机吧~相反,tdf文件很小,导入IGV不会出现反应慢,但是tdf文件只能反映基因组每个区域的测序深度,无法看到具体的比对情况,适合用来check找到的peak或者CNV(参考bam文件的可视化(测序深度) | IGV)。
(七)call peak
使用MACS2来进行call peak(MACS2官网:README for MACS (1.4.2))。Chip-seq与ATAC-seq call peaks的区别,前者peaks是代表抗体结合转录因子的位点,后者peaks是代表Tn5转座酶切开染色质开放区的两端,因此在一个位置,前者peaks有一个,后者有两个(参考:ATAC-seq 分析(上))。
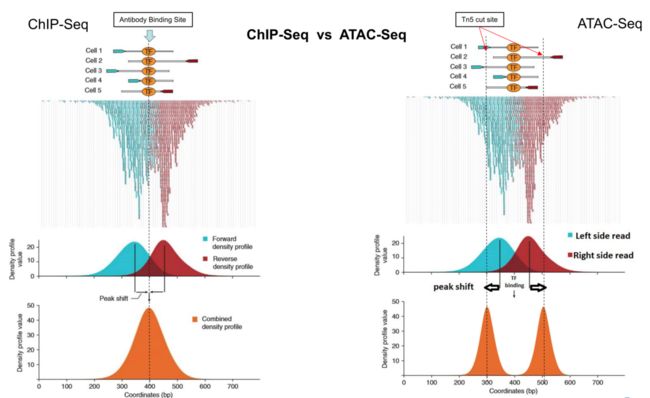
ATAC-seq关心的是在哪切断,断点才是peak的中心,所以使用shift模型(参考第3篇:用MACS2软件call peaks):
#!/bin/bash
names=$(ls /media/yanfang/FYWD/ATAC/sam_files/*.rmdup_sorted.bam)
for i in ${names}
do
macs2 callpeak -t $i -g mm --nomodel --shift -100 --extsize 200 -f BAM -p 1e-4 -n $i --outdir /media/yanfang/FYWD/ATAC/callpeak/ 2> $i.macs2.log
done
-t/–treatment: 输入文件,支持很多格式,搭配-f/–format使用。
-g/–gsize: 提供基因组的大小,程序有默认的几个物种可以选hs,mm,ce,dm。
--nomodel:该参数与extsize、shift配套使用。
--extsize:从5'->3'方向延伸reads。比如:你的转录因子结合范围200bp,就设置这个参数是200。
--shift:用这个参数从5' 端移动剪切,然后用--extsize延伸,如果--shift是负值表示从3'端方向移动。建议ChIP-seq数据集这个值保持默认值为0,对于检测富集剪切位点如DNAsel数据集设置为EXTSIZE的一半。
-f/–format: 设定输入文件的格式,这里选择bam格式。
-p/–pvalue: 设定pvalue的阈值,如果参数设定了-p,则其会覆盖参数-q的作用。
-n/–name: 输出文件(有很多个文件)的前缀。
运行之后,看一下每个样本找到多少个peak(bed的格式,储存了每个peaks的位置信息):
$ wc -l *.bed
33482 SCC_1_CD71hi_GFPhi.sam.filtered.bam.rmdup_sorted.bam_summits.bed
32267 SCC_1_CD71hi_GFPlo.sam.filtered.bam.rmdup_sorted.bam_summits.bed
35458 SCC_1_CD71lo_GFPlo.sam.filtered.bam.rmdup_sorted.bam_summits.bed
42029 SCC_1_CD71o_GFPhi.sam.filtered.bam.rmdup_sorted.bam_summits.bed
33667 SCC_2_CD71hi_GFPhi.sam.filtered.bam.rmdup_sorted.bam_summits.bed
20121 SCC_2_CD71hi_GFPlo.sam.filtered.bam.rmdup_sorted.bam_summits.bed
20287 SCC_2_CD71lo_GFPlo.sam.filtered.bam.rmdup_sorted.bam_summits.bed
35771 SCC_2_CD71o_GFPhi.sam.filtered.bam.rmdup_sorted.bam_summits.bed
(八)使用deeptools标准化
#!/bin/bash
tobw=$(ls /media/yanfang/FYWD/ATAC/bam_files/*.rmdup_sorted.bam)
for i in ${tobw}
do
bamCoverage -b $i -o $i.bin10.bw --binSize 10 -p 10 --normalizeUsing RPKM
done
NOTE:上面的参数说明请见:ChIP-seq实践(非转录因子,非组蛋白)
(九)IGV可视化
标准化后,会生成bw文件(bigwig文件),可直接导入IGV查看。现在bw文件,就可以在IGV里进行比较了,因为这些文件都是标准化之后的。

