拟时(pseudotime)分析,又称细胞轨迹(cell trajectory)分析,通过拟时分析可以推断出发育过程细胞的分化轨迹或细胞亚型的演化过程,在发育相关研究中使用频率较高。主要基于关键基因的表达模式,在拟时间中对单个细胞进行排序,模拟出时间发育过程的动态变化。
既然讲到所谓的拟时分析不过是一种排序而已,那么我们就要知道排序的基本要素:
- 对什么排序
- 如何判断先后顺序
- 如何寻找分支点(如果分支的话)
早在20世纪30年代,数量生态学领域前苏联学者Ranensky就提出了排序的概念,并发展了一个简单的排序方法(见Sobolev和Utekhin 1973),但只限于在前苏联传播(Greig-Smith 1980),Ramensky当时应用一个或两个环境因子梯度去排列植物群落,他用的名词是德文“ordnung”。经过多年的发展,排序技术依然是一种在低维空间排布高维数据的降维技术。当我们讲到排序的时候,离不开降维;讲到降维的时候,离不开特征提取(或者选择)。
那么对应到拟时分析的描述中:1)关键基因就是特征选择的结果,2)拟时间就是排序空间,3)排序就是细胞的演化轨迹。所有的拟时分析都离不开这三点。具体地:
Monocle引入了在伪时间(拟时间)内对单个细胞排序的策略,利用单个细胞的非同步进程,将它们置于与细胞分化等生物学过程相对应的轨迹上。Monocle利用先进的机器学习技术(反向图嵌入)从单细胞数据中学习显式的主图来对细胞进行排序,该方法能够可靠、准确地解决复杂的生物学过程。
Monocle2 可以做什么?
- Clustering, classifying, and counting cells
- Constructing single-cell trajectories.
- Differential expression analysis
当然我们关心的是第二个功能了,但是不防也看一下它的其他功能。算法原理可以参看文章Reversed graph embedding resolves complex single-cell developmental trajectories
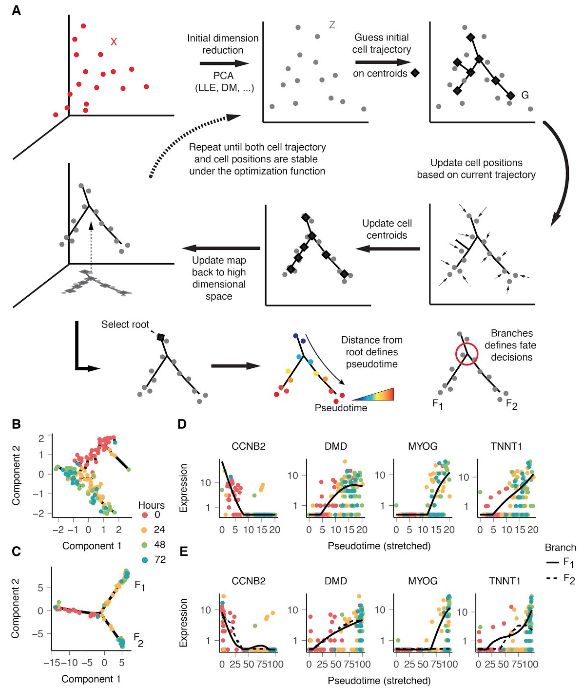
Monocle 2通过反向图嵌入学习单细胞轨迹的方法。每个细胞都可以表示为高维空间中的一个点,其中每个维对应于有序基因的表达水平。高维数据首先通过PCA(默认方法)、扩散图(diffusion maps)等几种降维方法中的任何一种投影到较低的维空间。然后Monocle 2在自动选择的数据中心集上构造一个生成树。然后,该算法细胞移动到树中最近的顶点,不断更新顶点的位置以“适合”细胞,学习新的生成树,并迭代地继续这个过程,直到树和细胞的位置收敛为止。在整个过程中,Monocle 2保持了高维空间和低维空间之间的可逆映射,从而既学习了轨迹,又降低了数据的维数。一旦Monocle 2习得这棵树,并选择一个tip作为“根”。每个细胞的伪时间计算为其沿树到根的测地距离(Geodesic Distance,在图论中,Geodesic Distance 就是图中两节点的最短路径的距离),并根据主图自动分配其分支。
主要过程如下,最难理解的应该是反转图嵌入,对算法感兴趣的同学可以研究一下流行学习的知识。
- dpFeature selection, identifying genes that define biological process 确定和生物过程有关的基因
selecting the genes differentially expressed between clusters of cells identified with tSNE dimension reduction followed by density peak clustering选择差异表达基因; - 反向图嵌入(GRE):默认DDRTree。它是一种流形学习算法,旨在将主要图形嵌入高维单细胞RNA-seq数据中,找到高维基因表达空间与低维空间映射,同时在这个缩小的空间中学习图的结构(K个质心)来解决这个问题;
- 伪时间分配和分支计算:利用DDRTree构建MST,将细胞投影到MST上,MST递归计算伪时间;
算法层面适可而止。
seurat2monocle
library(monocle)
#<-importCDS(pbmc,import_all = TRUE)
#Load Seurat object
pbmc <- readRDS('D:\\Users\\Administrator\\Desktop\\RStudio\\single_cell\\filtered_gene_bc_matrices\\hg19pbmc_tutorial.rds')
#Extract data, phenotype data, and feature data from the SeuratObject
data <- as(as.matrix(pbmc@assays$RNA@counts), 'sparseMatrix')
pd <- new('AnnotatedDataFrame', data = [email protected])
fData <- data.frame(gene_short_name = row.names(data), row.names = row.names(data))
fd <- new('AnnotatedDataFrame', data = fData)
#Construct monocle cds
monocle_cds <- newCellDataSet(data,
phenoData = pd,
featureData = fd,
lowerDetectionLimit = 0.5,
expressionFamily = negbinomial.size())
如果你只有count矩阵可以这么读入:
#do not run
HSMM_expr_matrix <- read.table("fpkm_matrix.txt")
HSMM_sample_sheet <- read.delim("cell_sample_sheet.txt")
HSMM_gene_annotation <- read.delim("gene_annotations.txt")
pd <- new("AnnotatedDataFrame", data = HSMM_sample_sheet)
fd <- new("AnnotatedDataFrame", data = HSMM_gene_annotation)
HSMM <- newCellDataSet(as.matrix(HSMM_expr_matrix),
phenoData = pd, featureData = fd)
这里重点是理解CellDataSet 的数据结构。
- exprs, a numeric matrix of expression values, where rows are genes, and columns are cells
- phenoData, an AnnotatedDataFrame object, where rows are cells, and columns are cell attributes** (such as cell type, culture condition, day captured, etc.)
- featureData, an AnnotatedDataFrame object, where rows are features (e.g. genes), and columns are gene attributes, such as biotype, gc content, etc.
官网给出许多其他建议以及读入数据的注意事项,需要注意的是,如果是UMI数据,不应该在创建CellDataSet之前自己对其进行标准化。也不应该试图将UMI计数转换为相对丰度(通过将其转换为FPKM/TPM数据)。Monocle将在内部执行所有需要的标准化步骤。自己将其正常化可能会破坏Monocle的一些关键步骤。
数据过滤(QC)
> HSMM<-monocle_cds
## 归一化
> HSMM <- estimateSizeFactors(HSMM)
> HSMM <- estimateDispersions(HSMM)
#Filtering low-quality cells
> HSMM <- detectGenes(HSMM, min_expr = 3 )
> print(head(fData(HSMM)))
gene_short_name num_cells_expressed
AL627309.1 AL627309.1 9
AP006222.2 AP006222.2 3
RP11-206L10.2 RP11-206L10.2 5
RP11-206L10.9 RP11-206L10.9 3
LINC00115 LINC00115 18
NOC2L NOC2L 254
> expressed_genes <- row.names(subset(fData(HSMM),
+ num_cells_expressed >= 10))
> print(head(pData(HSMM)))
orig.ident nCount_RNA nFeature_RNA percent.mt RNA_snn_res.0.5 seurat_clusters Size_Factor num_genes_expressed
AAACATACAACCAC pbmc3k 2419 779 3.0177759 1 1 NA 779
AAACATTGAGCTAC pbmc3k 4903 1352 3.7935958 3 3 NA 1352
AAACATTGATCAGC pbmc3k 3147 1129 0.8897363 1 1 NA 1129
AAACCGTGCTTCCG pbmc3k 2639 960 1.7430845 2 2 NA 960
AAACCGTGTATGCG pbmc3k 980 521 1.2244898 6 6 NA 521
AAACGCACTGGTAC pbmc3k 2163 781 1.6643551 1 1 NA 781
细胞分类(Classifying)
Monocle提供了一个简单的系统,可以根据您选择的marker基因的表达来标记细胞。您只需提供Monocle可以用来注释每个单元格的一组函数。
CDC20 <- row.names(subset(fData(HSMM), gene_short_name == "CDC20"))
GABPB2 <- row.names(subset(fData(HSMM),
gene_short_name == "GABPB2"))
cth <- newCellTypeHierarchy()
cth <- addCellType(cth, "CDC20", classify_func =
function(x) { x[CDC20,] >= 1 })
cth <- addCellType(cth, "GABPB2", classify_func = function(x){ x[GABPB2,] >= 1 })
HSMM <- classifyCells(HSMM, cth, 0.1)
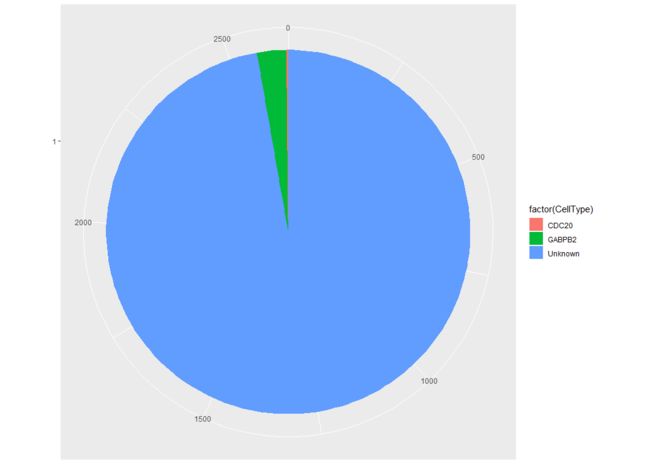
table(pData(HSMM)$CellType)
CDC20 GABPB2 Unknown
4 69 2565
我只是随意拿几个基因来试一下
pie <- ggplot(pData(HSMM),
aes(x = factor(1), fill = factor(CellType))) + geom_bar(width = 1)
pie + coord_polar(theta = "y") +
theme(axis.title.x = element_blank(), axis.title.y = element_blank())
聚类
第一步是决定用哪些基因来进行聚类(特征选择)。我们可以使用所有的基因,但是我们将包括很多没有表达到足够高水平来提供有意义的信号的基因,它们只会给系统增加噪音。我们可以根据平均表达水平筛选基因,我们还可以选择细胞间异常变异的基因。这些基因往往对细胞状态有很高的信息含量。
#Clustering cells without marker genes
disp_table <- dispersionTable(HSMM)
unsup_clustering_genes <- subset(disp_table, mean_expression >= 0.1)
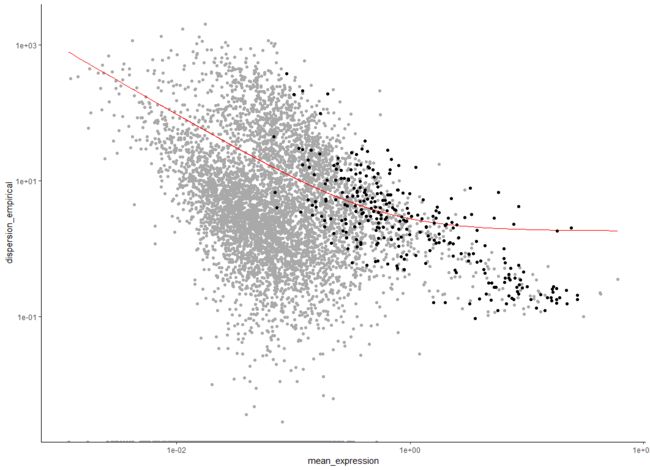
HSMM <- setOrderingFilter(HSMM, unsup_clustering_genes$gene_id)
plot_ordering_genes(HSMM)
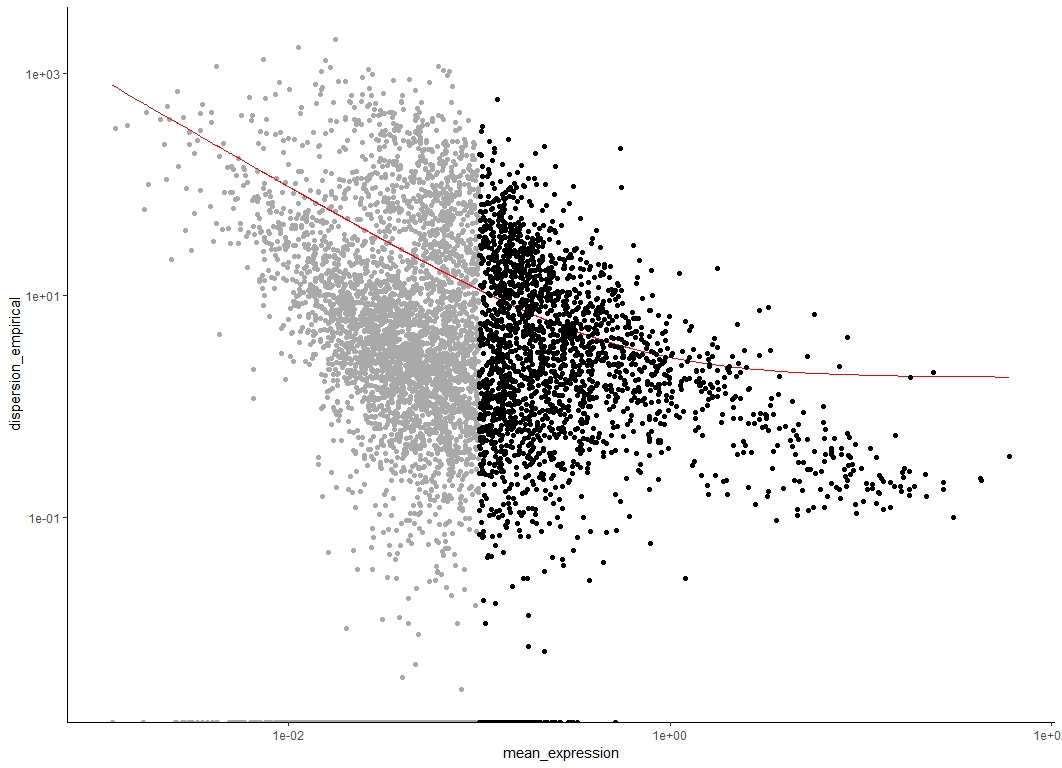
红线表示单片基于这种关系对色散的期望。我们标记用于聚类的基因用黑点表示,其他的用灰点表示。
# Plots the percentage of variance explained by the each component based on PCA from the normalized expression data using the same procedure used in reduceDimension function.
# HSMM@auxClusteringData[["tSNE"]]$variance_explained <- NULL
plot_pc_variance_explained(HSMM, return_all = F) # norm_method='log'
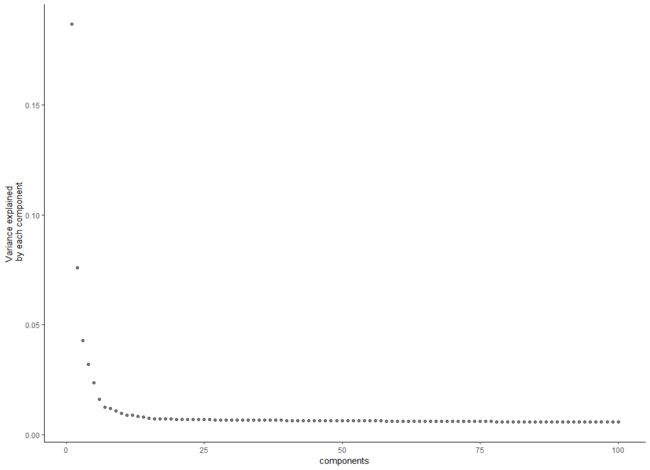
> HSMM <- reduceDimension(HSMM, max_components = 2, num_dim = 10,
+ reduction_method = 'tSNE', verbose = T)
Remove noise by PCA ...
Reduce dimension by tSNE ...
> HSMM <- clusterCells(HSMM, num_clusters = 2)
Distance cutoff calculated to 2.589424
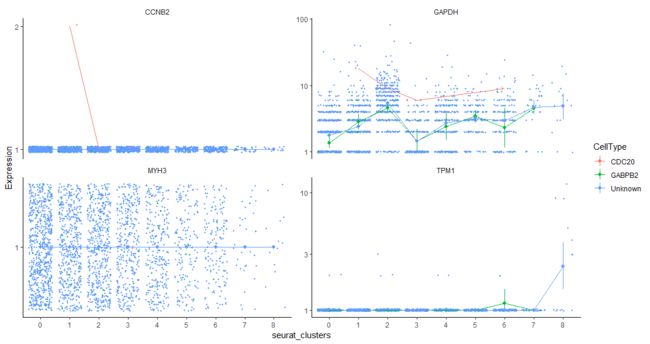
> plot_cell_clusters(HSMM, 1, 2, color = "CellType",
+ markers = c("CDC20", "GABPB2"))
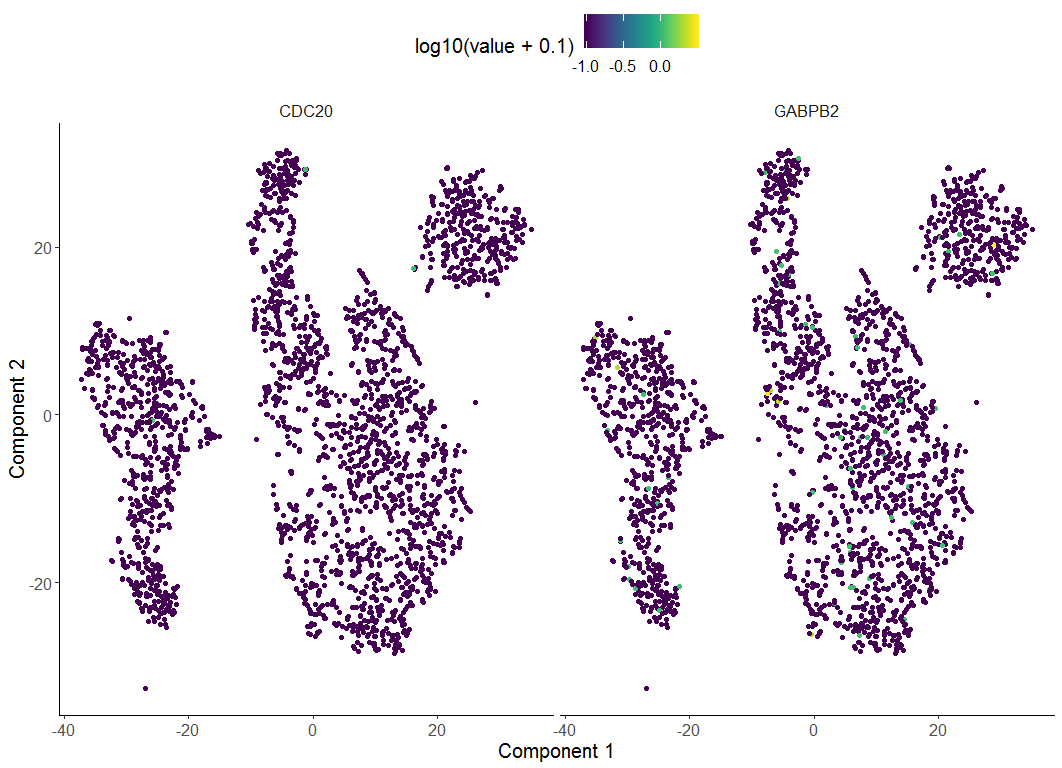
我发现指定cluster为5的时候,只能画出来4个,为什么?
HSMM <- clusterCells(HSMM, num_clusters = 5)
plot_cell_clusters(HSMM, 1, 2)
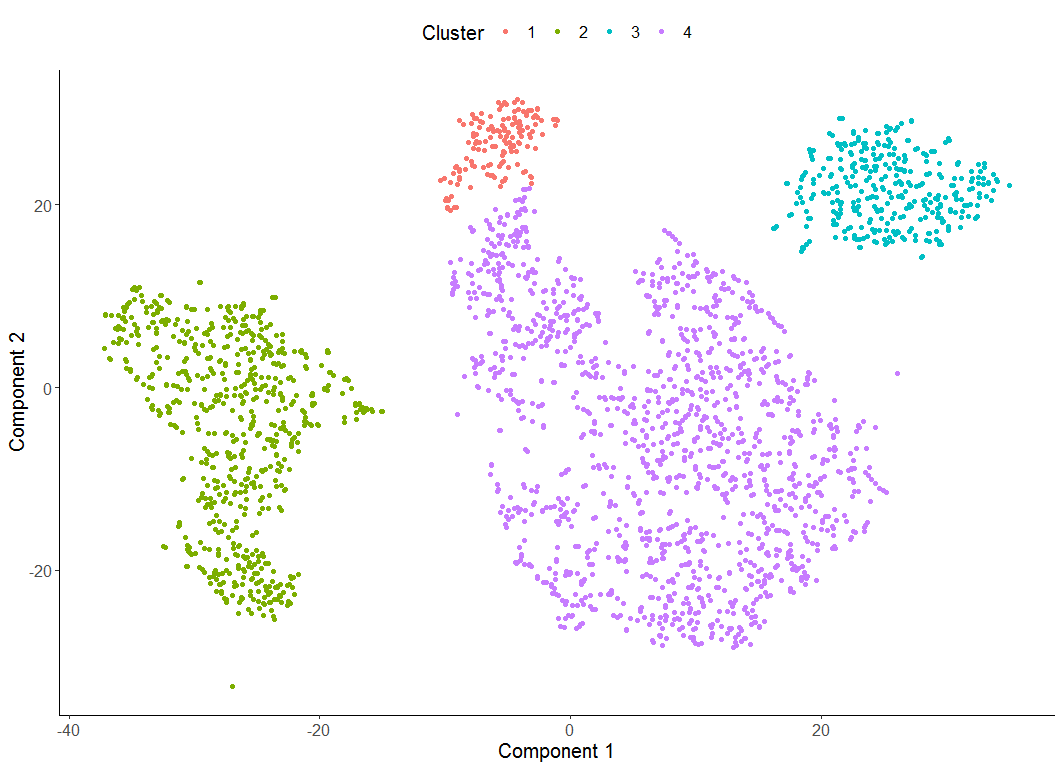
Monocle允许我们减去“无趣的”变异源的影响,以减少它们对集群的影响。您可以使用到clusterCells和其他几个Monocle函数的residualmodelformula astr参数来实现这一点。此参数接受一个R模型公式字符串,该字符串指定要在cluster之前减去。
HSMM <- reduceDimension(HSMM, max_components = 2, num_dim = 2,
reduction_method = 'tSNE',
residualModelFormulaStr = "~Size_Factor + num_genes_expressed",
verbose = T)
HSMM <- clusterCells(HSMM, num_clusters = 5)
plot_cell_clusters(HSMM, 1, 2, color = "Cluster") # 图不放了
plot_cell_clusters(HSMM, 1, 2, color = "Cluster") +
facet_wrap(~CellType)
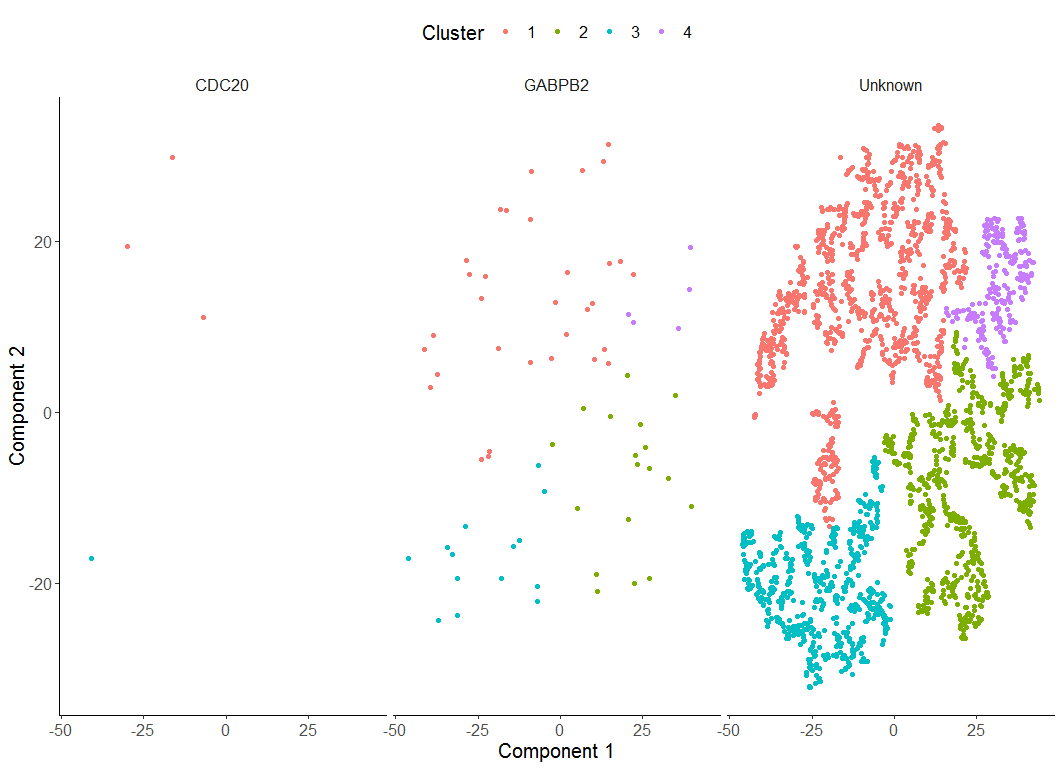
monocle 支持半监督聚类Clustering cells using marker genes来定义细胞以及外部导入细胞类型(Imputing cell type)(cell与type对应文件),这里均不再展示。
构建轨迹
在发育过程中,细胞会对刺激做出反应,并在整个生命过程中,从一种功能“状态”转变为另一种功能“状态”。不同状态的细胞表达不同的基因,产生蛋白质和代谢物的动态重复序列,从而完成它们的工作。当细胞在不同状态间移动时,会经历一个转录重组的过程,其中一些基因被沉默,另一些基因被激活。这些瞬时状态通常很难描述,因为在更稳定的端点状态之间纯化细胞可能是困难的或不可能的。
单细胞RNA-Seq可以使您在不需要纯化的情况下看到这些状态。然而,要做到这一点,我们必须确定每个细胞的可能状态区间。
Monocle介绍了利用RNA-Seq进行单细胞轨迹分析的策略。Monocle不是通过实验将细胞纯化成离散状态,而是使用一种算法来学习每个细胞必须经历的基因表达变化序列,作为动态生物学过程的一部分。一旦它了解了基因表达变化的整体“轨迹”,Monocle就可以将每个细胞置于轨迹中的适当位置。
然后,您可以使用Monocle的微分分析工具包来查找在轨迹过程中受到调控的基因,如查找作为伪时间函数变化的基因。如果这个过程有多个结果,Monocle将重建一个“分支”轨迹。这些分支与细胞的“决策”相对应,Monocle提供了强大的工具来识别受它们影响的基因,并参与这些基因的形成以及如何进行分析分支。Monocle依靠一种叫做反向图嵌入的机器学习技术来构建单细胞轨迹。
在开始之前您也需要问,什么是拟时(伪时间)分析。
伪时间是衡量单个细胞在细胞分化等过程中取得了多大进展的指标。在许多生物学过程中,细胞并不是完全同步的。在细胞分化等过程的单细胞表达研究中,捕获的细胞在分化方面可能分布广泛。也就是说,在同一时间捕获的细胞群中,有些细胞可能已经很长时间了,而有些细胞甚至还没有开始这个过程。
当您想要了解在细胞从一种状态转换到另一种状态时所发生的调节更改的顺序时,这种异步性会产生主要问题。跟踪同时捕获的细胞间的表达可以产生对基因动力学一个大致的认识,该基因表达的明显变异性将非常高。Monocle根据每个cell在学习轨迹上的进展对其进行排序,从而缓解了由于异步而产生的问题。
Monocle不是跟踪表达式随时间变化的函数,而是跟踪沿轨迹变化的函数,我们称之为伪时间。伪时间是一个抽象的分化单位:它只是一个cell到轨迹起点的距离,沿着最短路径测量。轨迹的总长度是由细胞从起始状态移动到结束状态所经历的总转录变化量来定义的。
The ordering workflow
- Step 1: choosing genes that define progress
- Step 2: reducing the dimensionality of the data
- Step 3: ordering the cells in pseudotime
基因的选择
首先,我们必须决定我们将使用哪些基因来定义细胞在肌生成过程中的进展。我们最终想要的是一组基因,能够随着我们研究过程的进展而增加(或减少)表达。
理想情况下,我们希望尽可能少地使用正在研究的系统生物学的先验知识。我们希望从数据中发现重要的排序基因,而不是依赖于文献和教科书,因为这可能会在排序中引入偏见。我们将从一种更简单的方法开始,但是我们通常推荐一种更复杂的方法,称为“dpFeature”。
diff_test_res <- differentialGeneTest(HSMM[expressed_genes,],
fullModelFormulaStr = "~percent.mt")
ordering_genes <- row.names (subset(diff_test_res, qval < 0.1)) ## 不要也写0.1 ,而是要写0.01。
HSMM <- setOrderingFilter(HSMM, ordering_genes)
plot_ordering_genes(HSMM)
根据时间点的差异分析来选择基因通常是非常有效的,但是如果我们没有时间序列数据呢?如果细胞在我们的生物过程中是异步分化的(通常是这样),Monocle通常可以从同时捕获的单个种群重建它们的轨迹。下面是两种选择基因的方法,它们完全不需要知道实验的设计。
一旦我们有了一个用于排序的基因id列表,我们就需要在HSMM对象中设置它们,因为接下来的几个函数将依赖于它们。
降维
#Trajectory step 2: reduce data dimensionality
HSMM <- reduceDimension(HSMM, max_components = 2,
method = 'DDRTree')
排序
#Trajectory step 3: order cells along the trajectory
HSMM <- orderCells(HSMM)
plot_cell_trajectory(HSMM, color_by = "seurat_clusters")
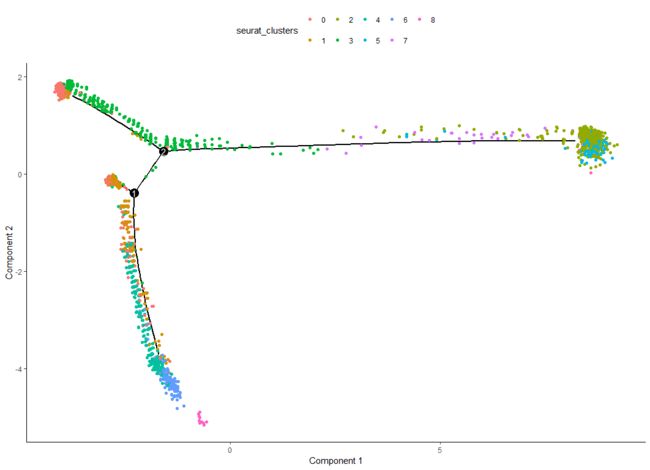
有了树结构分类颜色是可以侄自己指定的。轨迹呈树形结构,Monocle事先不知道应该调用树的哪个轨迹中的哪个来调用“开始”,所以我们经常不得不再次使用root_state参数调用orderCells来指定开始。首先,我们绘制轨迹,这次按“状态”给细胞上色。
···
plot_cell_trajectory(HSMM, color_by = "State")
···
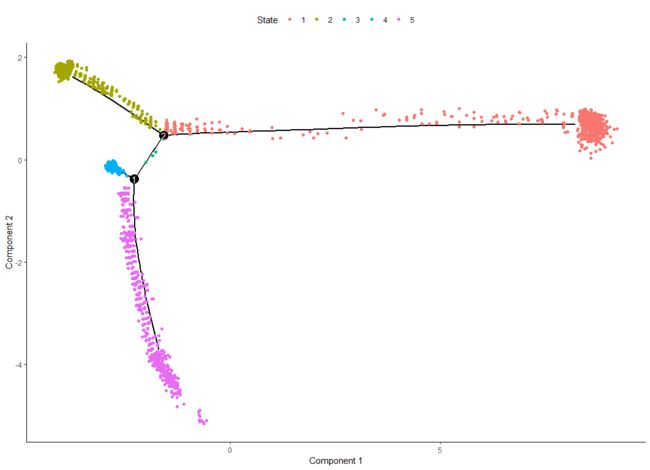
plot_cell_trajectory(HSMM, color_by = "Pseudotime")
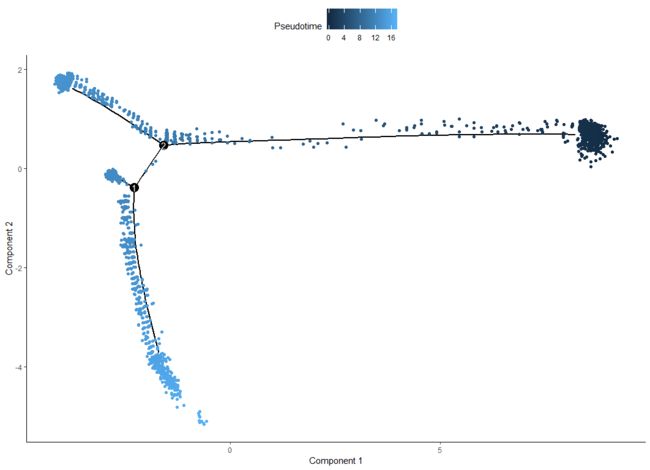
“状态”只是单片树的术语,表示这段树。下面的函数便于识别包含时间为零的大多数细胞的状态。然后我们可以把它传递给orderCells
GM_state <- function(cds){
if (length(unique(pData(cds)$State)) > 1){
T0_counts <- table(pData(cds)$State, pData(cds)$seurat_clusters)[,"0"]
return(as.numeric(names(T0_counts)[which
(T0_counts == max(T0_counts))]))
} else {
return (1)
}
}
HSMM_1 <- orderCells(HSMM, root_state = GM_state(HSMM))
plot_cell_trajectory(HSMM_1, color_by = "Pseudotime")
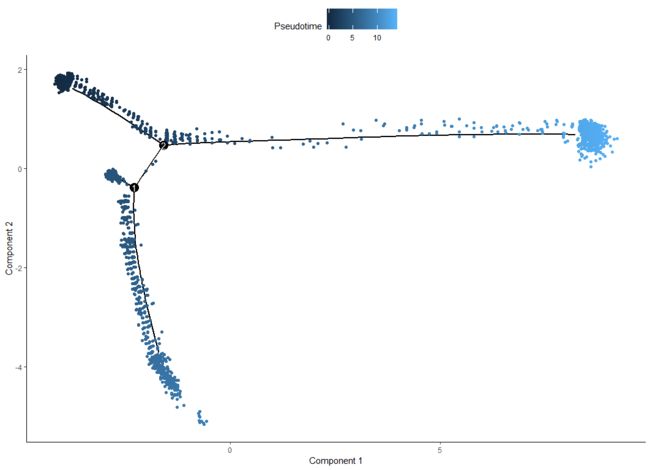
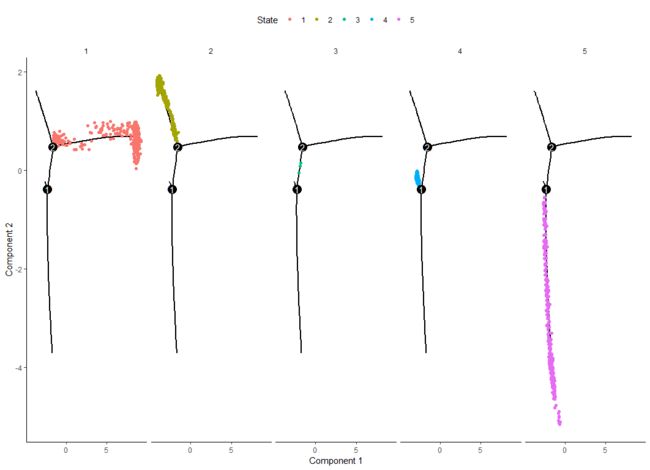
可以看出不同的排序条件拟时方向是不一样的。同时如果想看每个状态下细胞的分化可以指定分面,当然你也可以指定其他的分面方式。
plot_cell_trajectory(HSMM_myo, color_by = "State") +
facet_wrap(~State, nrow = 1)
如果没有timeseries,可能需要根据某些标记基因的表达位置来设置根,使用您对系统的生物学知识。例如,在这个实验中,高度增殖的祖细胞群产生两种有丝分裂后的细胞。所以根应该有表达高水平增殖标记的细胞。我们可以使用抖动图找出对应于快速增殖的状态
blast_genes <- row.names(subset(fData(HSMM),
gene_short_name %in% c("CCNB2", "NOC2L", "CDC20")))
plot_genes_jitter(HSMM[blast_genes,],
grouping = "State",
min_expr = 0.1)
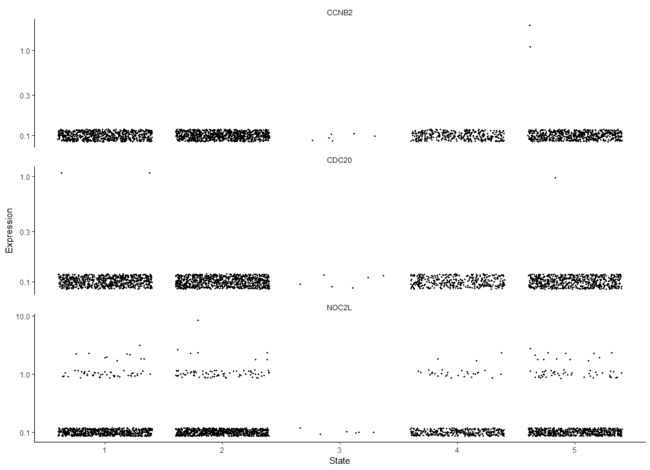
单个基因的时间变化(我可以随意选择基因而你不可以,你要选有意义的生活)
HSMM_expressed_genes <- row.names(subset(fData(HSMM),
num_cells_expressed >= 10))
HSMM_filtered <- HSMM[HSMM_expressed_genes,]
my_genes <- row.names(subset(fData(HSMM_filtered),
gene_short_name %in% c("YWHAB", "ICAM2", "ICAM2")))
cds_subset <- HSMM_filtered[my_genes,]
plot_genes_in_pseudotime(cds_subset, color_by = "seurat_clusters")
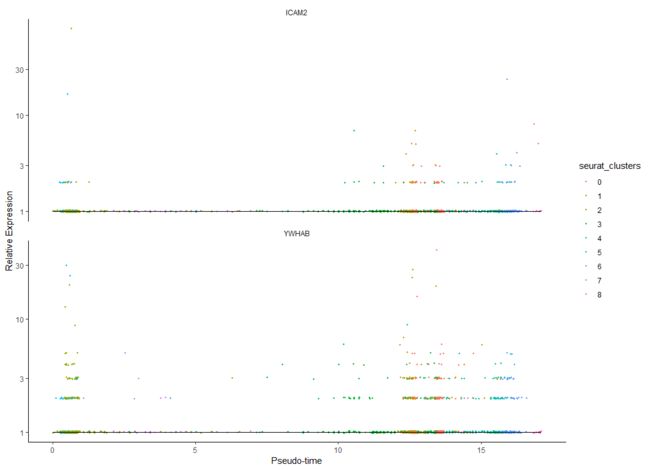
差异分析
差异基因表达分析是RNA-Seq实验中的一项常见任务。Monocle可以帮助你找到不同细胞群间差异表达的基因,并评估这些变化的统计显著性。这些比较要求您有一种方法将细胞收集到两个或更多组中。这些组由每个CellDataSet的表现型数据表中的列定义。Monocle将评估不同细胞群中每个基因表达水平的显著性。
marker_genes <- row.names(subset(fData(HSMM),
gene_short_name %in% c("MEF2C", "MEF2D", "MYF5",
"ANPEP", "PDGFRA","MYOG",
"TPM1", "TPM2", "MYH2",
"MYH3", "NCAM1", "TNNT1",
"TNNT2", "TNNC1", "CDK1",
"CDK2", "CCNB1", "CCNB2",
"CCND1", "CCNA1", "ID1")))
diff_test_res <- differentialGeneTest(HSMM[marker_genes,],
fullModelFormulaStr = "~percent.mt")
> head(diff_test_res)
status family pval qval gene_short_name num_cells_expressed use_for_ordering
MEF2D OK negbinomial.size 0.10544931 0.4590520 MEF2D 1 FALSE
CCNB1 OK negbinomial.size 0.45277970 0.7043240 CCNB1 1 FALSE
MEF2C OK negbinomial.size 0.28733226 0.5028315 MEF2C 2 FALSE
TPM2 OK negbinomial.size 0.94348541 0.9696949 TPM2 0 FALSE
CDK1 OK negbinomial.size 0.12768261 0.4590520 CDK1 0 FALSE
CCND1 OK negbinomial.size 0.04021331 0.4590520 CCND1 0 FALSE
MYOG_ID1 <- HSMM[row.names(subset(fData(HSMM),
gene_short_name %in% c("CCND1", "CCNB2"))),]
plot_genes_jitter(MYOG_ID1, grouping = "seurat_clusters", ncol= 2)
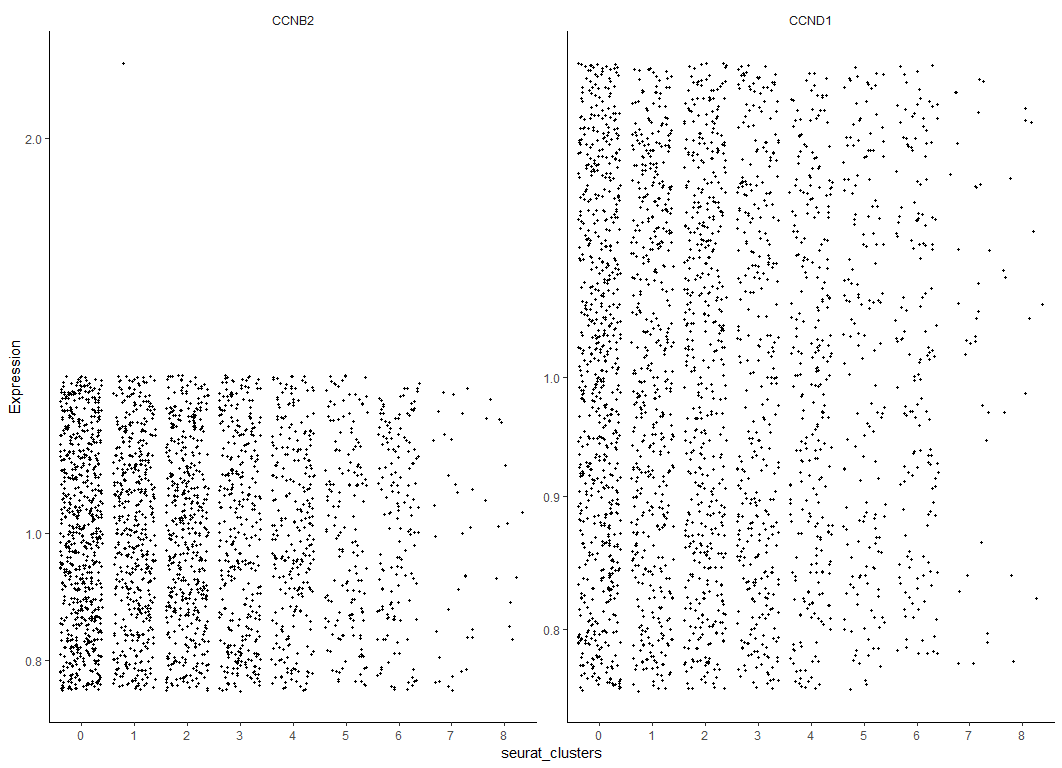
选择对区分细胞类型有意义的基因(Finding Genes that Distinguish Cell Type or State )
#Finding Genes that Distinguish Cell Type or State
to_be_tested <- row.names(subset(fData(HSMM),
gene_short_name %in% c("CDC20", "NCAM1", "ANPEP")))
cds_subset <- HSMM[to_be_tested,]
diff_test_res <- differentialGeneTest(cds_subset,
fullModelFormulaStr = "~CellType")
diff_test_res[,c("gene_short_name", "pval", "qval")]
plot_genes_jitter(cds_subset,
grouping = "CellType",
color_by = "CellType",
nrow= 1,
ncol = NULL,
plot_trend = TRUE)
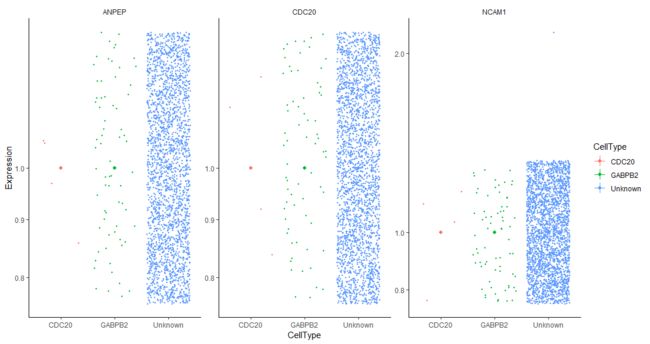
full_model_fits <-
fitModel(cds_subset, modelFormulaStr = "~CellType")
reduced_model_fits <- fitModel(cds_subset, modelFormulaStr = "~1")
diff_test_res <- compareModels(full_model_fits, reduced_model_fits)
diff_test_res
status family pval qval
CDC20 OK negbinomial.size 0.007822883 0.02346865
NCAM1 OK negbinomial.size 0.791131484 0.88906005
ANPEP OK negbinomial.size 0.889060052 0.88906005
Monocle中的差异分析过程非常灵活:您在测试中使用的模型公式可以包含pData表中作为列存在的任何项,包括Monocle在其他分析步骤中添加的列。例如,如果您使用集群细胞,您可以使用集群作为模型公式来测试cluster之间不同的基因。
Finding Genes that Change as a Function of Pseudotime
Monocle的主要工作是将细胞按照生物过程(如细胞分化)的顺序排列,而不事先知道要查看哪些基因。一旦这样做了,你就可以分析细胞,找到随着细胞进步而变化的基因。例如,你可以发现当细胞“成熟”时,基因显著上调。
to_be_tested <- row.names(subset(fData(HSMM),
gene_short_name %in% c("MYH3", "MEF2C", "CCNB2", "TNNT1")))
cds_subset <- HSMM[to_be_tested,]
diff_test_res <- differentialGeneTest(cds_subset,
fullModelFormulaStr = "~sm.ns(Pseudotime)")
diff_test_res[,c("gene_short_name", "pval", "qval")]
gene_short_name pval qval
MEF2C MEF2C 2.996841e-02 3.995789e-02
CCNB2 CCNB2 5.328721e-03 1.065744e-02
MYH3 MYH3 6.371156e-01 6.371156e-01
TNNT1 TNNT1 1.634704e-12 6.538818e-12
plot_genes_in_pseudotime(cds_subset, color_by = "seurat_clusters")
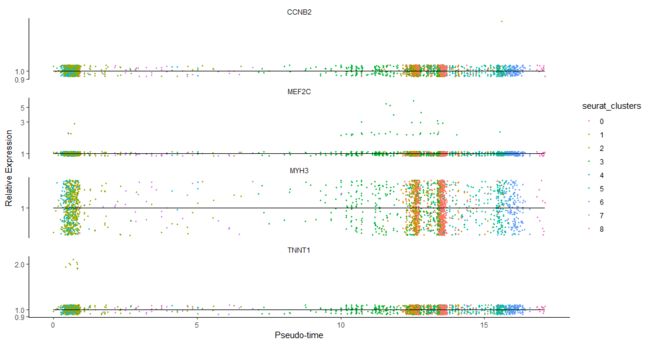
Clustering Genes by Pseudotemporal Expression Pattern
在研究时间序列基因表达研究时,一个常见的问题是:“哪些基因遵循相似的动力学趋势?”Monocle可以通过将具有相似趋势的基因分组来帮助你回答这个问题,这样你就可以分析这些基因组,看看它们有什么共同之处。Monocle提供了一种方便的方法来可视化所有伪时间相关的基因。函数plot_pseudotime_heatmap接受一个CellDataSet对象(通常只包含重要基因的子集),并生成与plot_genes_in_pseudotime类似的平滑表达曲线.然后,它将这些基因聚类并使用pheatmap包绘制它们。这允许您可视化基因模块,这些模块在伪时间内共同变化。
注意下面的num_clusters 指的是基因可以聚成几个类,而不是细胞。
diff_test_res <- differentialGeneTest(HSMM[marker_genes,],
fullModelFormulaStr = "~sm.ns(Pseudotime)")
sig_gene_names <- row.names(subset(diff_test_res, qval < 0.1))
plot_pseudotime_heatmap(HSMM[sig_gene_names,],
num_clusters = 6,
cores = 1,
show_rownames = T)
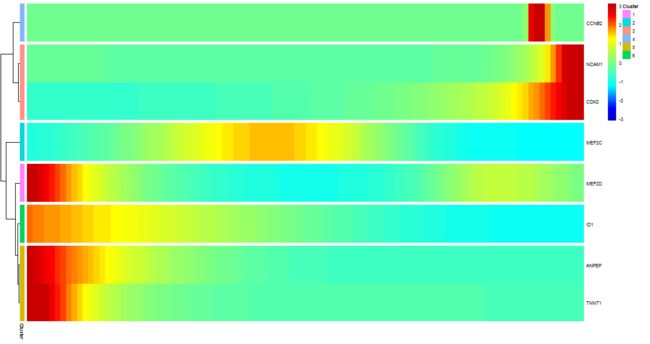
Multi-Factorial Differential Expression Analysis
Monocle可以在多个因素存在的情况下进行差异分析,这可以帮助你减去一些因素来看到其他因素的影响。在下面的简单例子中,Monocle测试了三个基因在成肌细胞和成纤维细胞之间的差异表达,同时减去percent.mt的影响。为此,我们必须同时指定完整模型和简化模型。完整的模型同时捕捉细胞类型和percent.mt的影响。
to_be_tested <-
row.names(subset(fData(HSMM),
gene_short_name %in% c("TPM1", "MYH3", "CCNB2", "GAPDH")))
cds_subset <- HSMM[to_be_tested,]
diff_test_res <- differentialGeneTest(cds_subset,
fullModelFormulaStr = "~CellType + percent.mt",
reducedModelFormulaStr = "~percent.mt")
diff_test_res[,c("gene_short_name", "pval", "qval")]
gene_short_name pval qval
GAPDH GAPDH 0.07990737 0.1598147
CCNB2 CCNB2 0.04148172 0.1598147
TPM1 TPM1 0.90861287 0.9086129
MYH3 MYH3 0.77085745 0.9086129
plot_genes_jitter(cds_subset,
grouping = "seurat_clusters", color_by = "CellType", plot_trend = TRUE) +
facet_wrap( ~ feature_label, scales= "free_y")
Analyzing Branches in Single-Cell Trajectories
通常,单细胞的轨迹包括分支。分支的产生是因为细胞执行替代基因表达程序。在发育过程中,当细胞做出命运的选择时,分支就会出现在轨迹中:一个发育谱系沿着一条路径前进,而另一个谱系产生第二条路径。Monocle包含用于分析这些分支事件的广泛功能。
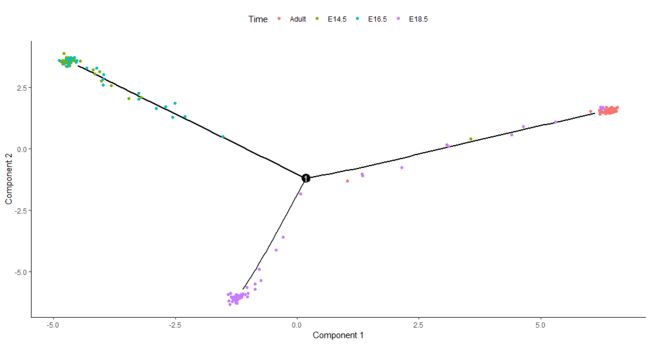
芭芭拉·特雷特琳和她的同事们在史蒂夫·奎克的实验室里进行了一项实验,他们从正在发育的老鼠肺中获取细胞。他们在细胞发育的早期捕获细胞,之后当肺包含两种主要类型的上皮细胞(AT1和AT2),以及即将决定成为AT1或AT2的细胞时。Monocle可以将这个过程重构为一个分支轨迹,让你可以非常详细地分析决策点。下图显示了使用Monocle的一些数据重建的一般方法。有一个单独的分支,标记为“1”。当细胞从树的左上方通过树枝的早期发育阶段通过时,哪些基因发生了变化?哪些基因在分支间有差异表达?为了回答这个问题,Monocle为您提供了一个特殊的统计测试:分支表达式分析建模,或BEAM。
lung <- load_lung()
plot_cell_trajectory(lung, color_by = "Time")
# 图如下
BEAM_res <- BEAM(lung, branch_point = 1, cores = 1)
BEAM_res <- BEAM_res[order(BEAM_res$qval),]
BEAM_res <- BEAM_res[,c("gene_short_name", "pval", "qval")]
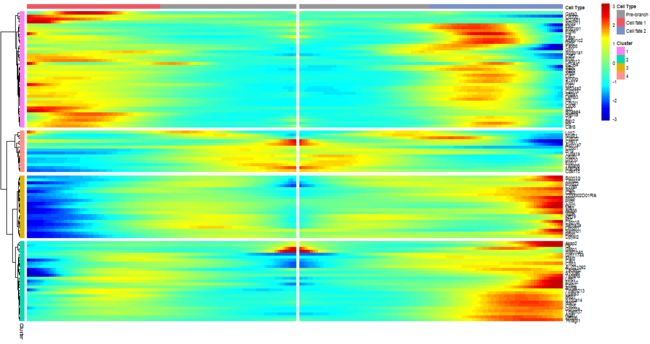
使用一种特殊类型的热图,您可以可视化所有明显依赖于分支的基因的变化。这张热图同时显示了两种血统的变化。它还要求您选择要检查的分支点。列是伪时间中的点,行是基因,伪时间的开始在热图的中间。当你从热图的中间读到右边的时候,你正在跟随一个伪时间谱系。当你读到左边时,另一个。这些基因是分层聚类的,因此您可以可视化具有类似的依赖于序列的基因模块.
plot_genes_branched_heatmap(lung[row.names(subset(BEAM_res,
qval < 1e-4)),],
branch_point = 1,
num_clusters = 4,
cores = 1,
use_gene_short_name = T,
show_rownames = T)
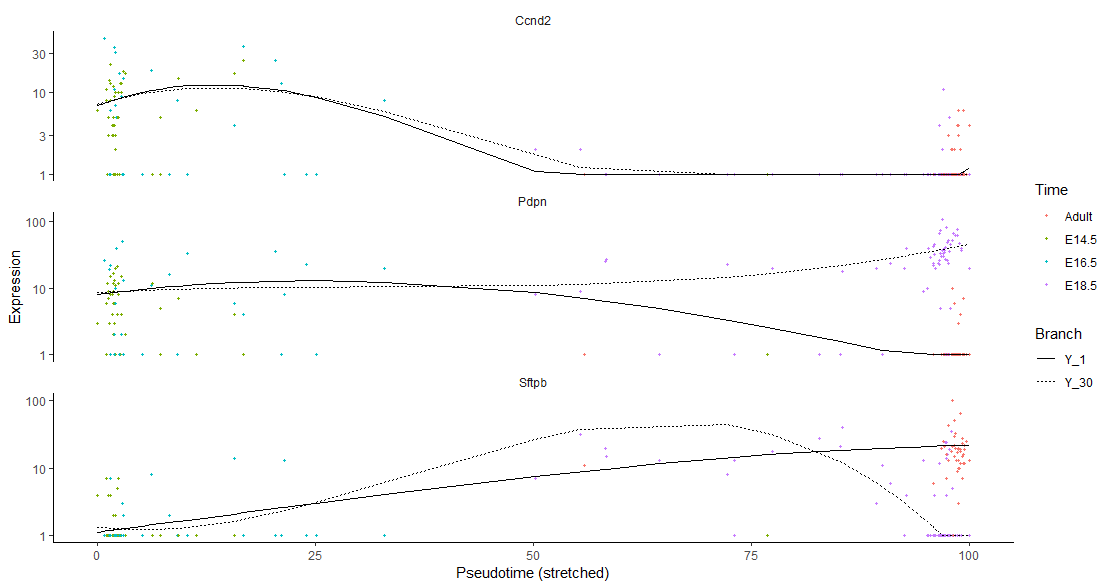
lung_genes <- row.names(subset(fData(lung),
gene_short_name %in% c("Ccnd2", "Sftpb", "Pdpn")))
plot_genes_branched_pseudotime(lung[lung_genes,],
branch_point = 1,
color_by = "Time",
ncol = 1)
> str(HSMM)
Formal class 'CellDataSet' [package "monocle"] with 19 slots
..@ reducedDimS : num [1:2, 1:2638] -2.613 -0.652 -1.784 0.745 -2.376 ...
.. ..- attr(*, "dimnames")=List of 2
.. .. ..$ : NULL
.. .. ..$ : chr [1:2638] "AAACATACAACCAC" "AAACATTGAGCTAC" "AAACATTGATCAGC" "AAACCGTGCTTCCG" ...
..@ reducedDimW : num [1:281, 1:2] -0.0384 0.0812 -0.0465 0.0681 0.0655 ...
..@ reducedDimA : num [1:2, 1:2638] 13.57 29.4 -2.74 -25.16 -9.3 ...
..@ reducedDimK : num [1:2, 1:126] -2.305 -0.692 -1.711 0.538 -1.935 ...
.. ..- attr(*, "dimnames")=List of 2
.. .. ..$ : NULL
.. .. ..$ : chr [1:126] "Y_1" "Y_2" "Y_3" "Y_4" ...
..@ minSpanningTree :List of 10
.. ..$ :List of 1
.. .. ..$ Y_1: 'igraph.vs' Named int [1:2] 47 101
.. .. .. ..- attr(*, "names")= chr [1:2] "Y_47" "Y_101"
.. .. .. ..- attr(*, "env")=
.. .. .. ..- attr(*, "graph")= chr "484d230c-94de-11e9-9f7f-07844984f7db"
.. ..$ :List of 1
.. .. ..$ Y_2: 'igraph.vs' Named int [1:2] 62 90
.. .. .. ..- attr(*, "names")= chr [1:2] "Y_62" "Y_90"
.. .. .. ..- attr(*, "env")=
.. .. .. ..- attr(*, "graph")= chr "484d230c-94de-11e9-9f7f-07844984f7db"
.. ..$ :List of 1
.. .. ..$ Y_3: 'igraph.vs' Named int [1:2] 90 126
.. .. .. ..- attr(*, "names")= chr [1:2] "Y_90" "Y_126"
.. .. .. ..- attr(*, "env")=
.. .. .. ..- attr(*, "graph")= chr "484d230c-94de-11e9-9f7f-07844984f7db"
.. ..$ :List of 1
.. .. ..$ Y_4: 'igraph.vs' Named int 106
.. .. .. ..- attr(*, "names")= chr "Y_106"
.. .. .. ..- attr(*, "env")=
.. .. .. ..- attr(*, "graph")= chr "484d230c-94de-11e9-9f7f-07844984f7db"
.. ..$ :List of 1
.. .. ..$ Y_5: 'igraph.vs' Named int [1:2] 36 65
.. .. .. ..- attr(*, "names")= chr [1:2] "Y_36" "Y_65"
.. .. .. ..- attr(*, "env")=
.. .. .. ..- attr(*, "graph")= chr "484d230c-94de-11e9-9f7f-07844984f7db"
.. ..$ :List of 1
.. .. ..$ Y_6: 'igraph.vs' Named int [1:2] 39 53
.. .. .. ..- attr(*, "names")= chr [1:2] "Y_39" "Y_53"
.. .. .. ..- attr(*, "env")=
.. .. .. ..- attr(*, "graph")= chr "484d230c-94de-11e9-9f7f-07844984f7db"
.. ..$ :List of 1
.. .. ..$ Y_7: 'igraph.vs' Named int [1:2] 34 98
.. .. .. ..- attr(*, "names")= chr [1:2] "Y_34" "Y_98"
.. .. .. ..- attr(*, "env")=
.. .. .. ..- attr(*, "graph")= chr "484d230c-94de-11e9-9f7f-07844984f7db"
.. ..$ :List of 1
.. .. ..$ Y_8: 'igraph.vs' Named int [1:2] 49 126
.. .. .. ..- attr(*, "names")= chr [1:2] "Y_49" "Y_126"
.. .. .. ..- attr(*, "env")=
.. .. .. ..- attr(*, "graph")= chr "484d230c-94de-11e9-9f7f-07844984f7db"
.. ..$ :List of 1
.. .. ..$ Y_9: 'igraph.vs' Named int [1:2] 42 93
.. .. .. ..- attr(*, "names")= chr [1:2] "Y_42" "Y_93"
.. .. .. ..- attr(*, "env")=
.. .. .. ..- attr(*, "graph")= chr "484d230c-94de-11e9-9f7f-07844984f7db"
.. ..$ :List of 1
.. .. ..$ Y_10: 'igraph.vs' Named int [1:2] 11 25
.. .. .. ..- attr(*, "names")= chr [1:2] "Y_11" "Y_25"
.. .. .. ..- attr(*, "env")=
.. .. .. ..- attr(*, "graph")= chr "484d230c-94de-11e9-9f7f-07844984f7db"
.. ..- attr(*, "class")= chr "igraph"
..@ cellPairwiseDistances: num [1:126, 1:126] 0 1.37 1.41 10.62 5.52 ...
.. ..- attr(*, "dimnames")=List of 2
.. .. ..$ : chr [1:126] "Y_1" "Y_2" "Y_3" "Y_4" ...
.. .. ..$ : chr [1:126] "Y_1" "Y_2" "Y_3" "Y_4" ...
..@ expressionFamily :Formal class 'vglmff' [package "VGAM"] with 22 slots
.. .. ..@ blurb : chr [1:6] "Negative-binomial distribution with size known\n\n" "Links: " "loglink(mu)" "\n" ...
.. .. ..@ constraints : expression({ constraints <- cm.zero.VGAM(constraints, x = x, NULL, M = M, predictors.names = predictors.names, M1 = 2) })
.. .. ..@ deviance :function ()
.. .. ..@ fini : expression({ })
.. .. ..@ first : expression({ })
.. .. ..@ infos :function (...)
.. .. ..@ initialize : expression({ M1 <- 1 temp5 <- w.y.check(w = w, y = y, Is.nonnegative.y = TRUE, Is.integer.y = TRUE, ncol.w.max | __truncated__
.. .. ..@ last : expression({ misc$link <- rep_len("loglink", NOS) names(misc$link) <- mynames1 misc$earg <- vector("list", M) | __truncated__
.. .. ..@ linkfun :function ()
.. .. ..@ linkinv :function (eta, extra = NULL)
.. .. ..@ loglikelihood :function (mu, y, w, residuals = FALSE, eta, extra = NULL, summation = TRUE)
.. .. ..@ middle : expression({ })
.. .. ..@ middle2 : expression({ })
.. .. ..@ summary.dispersion: logi(0)
.. .. ..@ vfamily : chr "negbinomial.size"
.. .. ..@ validparams :function (eta, y, extra = NULL)
.. .. ..@ validfitted :function ()
.. .. ..@ simslot :function (object, nsim)
.. .. ..@ hadof :function ()
.. .. ..@ charfun :function ()
.. .. ..@ deriv : expression({ eta <- cbind(eta) M <- ncol(eta) kmat <- matrix(Inf, n, M, byrow = TRUE) newemu <- list(theta = | __truncated__
.. .. ..@ weight : expression({ ned2l.dmunb2 <- 1/mu - 1/(mu + kmat) wz <- ned2l.dmunb2 * dmu.deta^2 c(w) * wz })
..@ lowerDetectionLimit : num 1
..@ dispFitInfo :
..@ dim_reduce_type : chr "DDRTree"
..@ auxOrderingData :
..@ auxClusteringData :
..@ experimentData :Formal class 'MIAME' [package "Biobase"] with 13 slots
.. .. ..@ name : chr ""
.. .. ..@ lab : chr ""
.. .. ..@ contact : chr ""
.. .. ..@ title : chr ""
.. .. ..@ abstract : chr ""
.. .. ..@ url : chr ""
.. .. ..@ pubMedIds : chr ""
.. .. ..@ samples : list()
.. .. ..@ hybridizations : list()
.. .. ..@ normControls : list()
.. .. ..@ preprocessing : list()
.. .. ..@ other : list()
.. .. ..@ .__classVersion__:Formal class 'Versions' [package "Biobase"] with 1 slot
.. .. .. .. ..@ .Data:List of 2
.. .. .. .. .. ..$ : int [1:3] 1 0 0
.. .. .. .. .. ..$ : int [1:3] 1 1 0
..@ assayData :
..@ phenoData :Formal class 'AnnotatedDataFrame' [package "Biobase"] with 4 slots
.. .. ..@ varMetadata :'data.frame': 17 obs. of 1 variable:
.. .. .. ..$ labelDescription: chr [1:17] NA NA NA NA ...
.. .. ..@ data :'data.frame': 2638 obs. of 17 variables:
.. .. .. ..$ orig.ident : Factor w/ 1 level "pbmc3k": 1 1 1 1 1 1 1 1 1 1 ...
.. .. .. ..$ nCount_RNA : num [1:2638] 2419 4903 3147 2639 980 ...
.. .. .. ..$ nFeature_RNA : int [1:2638] 779 1352 1129 960 521 781 782 790 532 550 ...
.. .. .. ..$ percent.mt : num [1:2638] 3.02 3.79 0.89 1.74 1.22 ...
.. .. .. ..$ RNA_snn_res.0.5 : Factor w/ 9 levels "0","1","2","3",..: 2 4 2 3 7 2 5 5 1 6 ...
.. .. .. ..$ seurat_clusters : Factor w/ 9 levels "0","1","2","3",..: 2 4 2 3 7 2 5 5 1 6 ...
.. .. .. ..$ Size_Factor : num [1:2638] 1.108 2.245 1.441 1.208 0.449 ...
.. .. .. ..$ num_genes_expressed : int [1:2638] 126 181 149 153 34 103 104 112 64 51 ...
.. .. .. ..$ Cluster : Factor w/ 1 level "1": 1 1 1 1 1 1 1 1 1 1 ...
.. .. .. ..$ peaks : logi [1:2638] FALSE FALSE FALSE FALSE FALSE FALSE ...
.. .. .. ..$ halo : logi [1:2638] FALSE FALSE FALSE FALSE FALSE FALSE ...
.. .. .. ..$ delta : num [1:2638] 0.401 0.442 1.031 0.205 0.812 ...
.. .. .. ..$ rho : num [1:2638] 36 33.3 44.8 47.7 31.7 ...
.. .. .. ..$ nearest_higher_density_neighbor: logi [1:2638] NA NA NA NA NA NA ...
.. .. .. ..$ Pseudotime : num [1:2638] 12.277 11.115 13.023 0.723 16.159 ...
.. .. .. ..$ State : Factor w/ 5 levels "1","2","3","4",..: 5 2 5 1 5 4 5 5 5 1 ...
.. .. .. ..$ CellType : Factor w/ 3 levels "CDC20","GABPB2",..: 3 3 3 3 3 3 3 3 3 3 ...
.. .. ..@ dimLabels : chr [1:2] "sampleNames" "sampleColumns"
.. .. ..@ .__classVersion__:Formal class 'Versions' [package "Biobase"] with 1 slot
.. .. .. .. ..@ .Data:List of 1
.. .. .. .. .. ..$ : int [1:3] 1 1 0
..@ featureData :Formal class 'AnnotatedDataFrame' [package "Biobase"] with 4 slots
.. .. ..@ varMetadata :'data.frame': 3 obs. of 1 variable:
.. .. .. ..$ labelDescription: chr [1:3] NA NA NA
.. .. ..@ data :'data.frame': 13714 obs. of 3 variables:
.. .. .. ..$ gene_short_name : Factor w/ 13714 levels "7SK.2","A1BG",..: 527 715 9394 9395 5971 7345 5750 8158 9747 4917 ...
.. .. .. ..$ num_cells_expressed: int [1:13714] 0 0 0 0 0 1 0 0 0 6 ...
.. .. .. ..$ use_for_ordering : logi [1:13714] FALSE FALSE FALSE FALSE FALSE FALSE ...
.. .. ..@ dimLabels : chr [1:2] "featureNames" "featureColumns"
.. .. ..@ .__classVersion__:Formal class 'Versions' [package "Biobase"] with 1 slot
.. .. .. .. ..@ .Data:List of 1
.. .. .. .. .. ..$ : int [1:3] 1 1 0
..@ annotation : chr(0)
..@ protocolData :Formal class 'AnnotatedDataFrame' [package "Biobase"] with 4 slots
.. .. ..@ varMetadata :'data.frame': 0 obs. of 1 variable:
.. .. .. ..$ labelDescription: chr(0)
.. .. ..@ data :'data.frame': 2638 obs. of 0 variables
.. .. ..@ dimLabels : chr [1:2] "sampleNames" "sampleColumns"
.. .. ..@ .__classVersion__:Formal class 'Versions' [package "Biobase"] with 1 slot
.. .. .. .. ..@ .Data:List of 1
.. .. .. .. .. ..$ : int [1:3] 1 1 0
..@ .__classVersion__ :Formal class 'Versions' [package "Biobase"] with 1 slot
.. .. ..@ .Data:List of 5
.. .. .. ..$ : int [1:3] 3 5 2
.. .. .. ..$ : int [1:3] 2 42 0
.. .. .. ..$ : int [1:3] 1 3 0
.. .. .. ..$ : int [1:3] 1 0 0
.. .. .. ..$ : int [1:3] 1 2 0
STREAM:一款scRNA-seq拟时分析工具
Clustering by fast search and find of density peaks
发表在 Science 上的一种新聚类算法
重磅消息:诺禾致源10X Genomics单细胞转录组产品全新升级
基迪奥10x单细胞转录组产品新升级
An overview of algorithms for estimating pseudotime in single-cell RNA-seq data
A set of tools supporting the development, execution, and benchmarking of trajectory inference methods.
hemberg-lab.github.io
Diffusion pseudotime robustly reconstructs lineage branching
dynverse
Monocle
A new statistical method allows researchers to infer different developmental processes from RNA-Seq data
Pseudotime estimation: deconfounding single cell time series
Your top 3 single-cell RNA sequencing analysis tools
Single-cell trajectory analysis
STREAM
MIA: Luca Pinello, Huidong Chen, Single-cell trajectories from omics; Jonathan Hsu, CRISPR tiling
使用monocle包进行pseudotime分析
关于细胞分化轨迹学习小笔记