复习资料:
表观调控13张图之一证明基因干扰有效性
表观调控13张图之二相关性热图看不同样本相关性
表观调控13张图之三。。。
在10月份的时候跟着学习了表观调控相关的联合组学分析,当时做了点记录,但是很多东西还是不熟悉,这次跟着公众号开始复习。
此次表观调控课程是整合RNA-seq数据和表观数据,比如Chip-seq和ATAC-seq,主要是依托于文章 Global changes of H3K27me3 domains and Polycomb group protein distribution in the absence of recruiters Spps or Pho 文献解读在 在果蝇探索 PRC 复合物(逆向收费读文献 2019-18) 这一推文。
原文链接:https://www.pnas.org/content/115/8/E1839
先回顾一下背景知识:
H3K27me3 是发生在组蛋白 H3 的第 27 位赖氨酸上的三甲基化修饰,从整体染色质水平来讲, 大量的证据支持 H3K27me3 与异染色质相关;从个体的基因水平来讲, H3K27me3 常常与转录抑制相关。H3K27me2/me3 甲基化的加载是由 PcG 复合体催化的。
如何精确地募集 Polycomb group(PcG)蛋白到其靶基因仍知之甚少。在果蝇中,PcG蛋白被募集到由多个 DNA 结合蛋白的结合位点组成的 Polycomb 反应元件( PRE )。为了了解如何将 PcG 蛋白募集到 PRE 并在其上维持,作者系统地研究了野生型的 PcG 结合,相关的 H3K27me3 和转录组以及三种 PRE 结合蛋白的突变体( Pho、Spps、cg )。
仅 Pho 结合位点不足以招募 PcG。PRE 由复杂的 DNA 结合蛋白的结合位点组成,包括Pho / Phol,Spps,Cg,GAF / Psq,Adf-1,Grh,Dsp1和Zeste / Fsh-S。然而这些蛋白质在 PcG 蛋白质募集中的作用尚不清楚。Spps 是与 PREs 结合的 Sp1 / KLF 锌指蛋白家族的成员
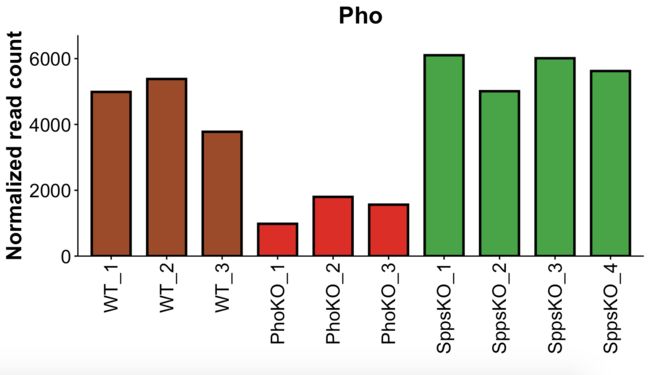
第一张图:说明基因干扰的有效性
当 PcG 招募成员被中断后,基因表达是如何改变的。进一步探索差异表达与 H3K27me3 修饰的改变有何关系?
作者对野生型、Spps 和 Pho 突变体 3 龄幼虫进行了 RNA-seq 测序。而本张图用来证实相应突变体中 Spps 和 Pho 表达确实降低了。
注: 当在检测时发现基因突变体的 RNA-seq 结果发生改变的时候,首先在获得表达量时候,第一件事情是去查看这些数据提示的基因在突变体中是否真的被抑制或者不表达了。
加载本次分析所涉及的包
rm(list = ls())
library(ggpubr)
library(stringr)
library(ggplot2)
library(cowplot)
自定义 barplot 主题
定义过的主题以后还可以重复使用
my_bar_theme <- theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5, size = 16),
# 因为 x 轴标签要旋转 90°,所以这里用来旋转
axis.text.y = element_text(size = 16),
axis.title.y = element_text(size = 18,
face = "bold",),
legend.position = "none") +
theme(plot.title = element_text(size = 20, face = "bold", hjust = 0.5)) # 基因名要居中,这里用来居中。
数据读取
options(stringsAsFactors = F)
a <- read.table('~/Downloads/fly/RNA-seq/counts/all.counts.id.txt', header = T)
# 查看数据行列信息,大致看下多少基因
dim(a)
> dim(a)
[1] 17714 16
因为我们的数据来源是一样的,所有结果也应该一样,由于存放的位置不同,路径做修改即可。
数据清洗、可视化
这一步非常重要,而且有几个难点,首先是获取数据,然后是排序,最后是取色,这里前辈用了Takecolor 软件进行对准样图取色,我是不会的,下去还需要学习一下,这里以及后面都可以可以参考曾老师的御用视频剪辑师的方法来完成。
链接:生信 | IGV导出svg文件后期美化
# 提取 `pho` 基因对应的行以及表达量信息
cg <- a[a[,1] == 'pho',7:16]
# 构建新的数据框以便进行可视化
dat <- data.frame(gene = as.numeric(cg),
sample = str_split(names(cg),'\\.',simplify = T)[,1],
group = str_split(names(cg),'_',simplify = T)[,1]
)
# 重定义 x 轴标签顺序
labels = c(paste0("WT", "_",1:3), paste0("PhoKO", "_", 1:3), paste0("SppsKO", "_", 1:4))
# 然后使用 `ggbarplot()` 函数进行绘图,其实我个人更喜欢用 `ggplot2` 来绘制,这个包也是基于 `ggplot2` 来的,所以对于我来说没啥区别,反而还变麻烦了。
ggbarplot(dat,x = 'sample', y = 'gene',
color = 'black', fill = "group",
size = 1) +
# 使用 Takecolor 软件进行对准样图取色
scale_fill_manual(values = c(WT = "#9C4B25",
PhoKO = "#DE2C1C",
SppsKO = "#43A542")) +
scale_color_manual(values = "black") +
scale_x_discrete(limits = labels) +
labs(y = "Normalized read count",
x = "",
title = "Pho") +
my_bar_theme +
scale_y_continuous(expand = c(0, 0)) +
geom_text(aes(y = gene * 1.1, label = ""))
封装画图函数
为了方便以后调用,因为要画多个基因的图来展示(也是偷懒的一种方式,哈哈)
my_barplot <- function(gene){
cg <- a[a[,1] == gene, 7:16] # 提取候选的表达量对应的行和列
dat <- data.frame(gene = as.numeric(cg),
sample = str_split(names(cg),'\\.',simplify = T)[,1], # 这里将样品后面的 bam文件后缀去掉
group = str_split(names(cg),'_',simplify = T)[,1]
)
# x 轴标签的顺序,这里是按照原图的顺序来的
labels = c(paste0("WT", "_",1:3), paste0("PhoKO", "_", 1:3), paste0("SppsKO", "_", 1:4))
ggbarplot(dat, x = 'sample', y = 'gene',
color = 'black', fill = "group",
size = 1) +
scale_fill_manual(values = c(WT = "#9C4B25",
PhoKO = "#DE2C1C",
SppsKO = "#43A542")) +
scale_color_manual(values = "black") +
scale_x_discrete(limits = labels) +
labs(y = "Normalized read count",
x = "",
title = gene) +
scale_y_continuous(expand = c(0, 0)) +
geom_text(aes(y = gene * 1.1, label = "")) +
my_bar_theme
}
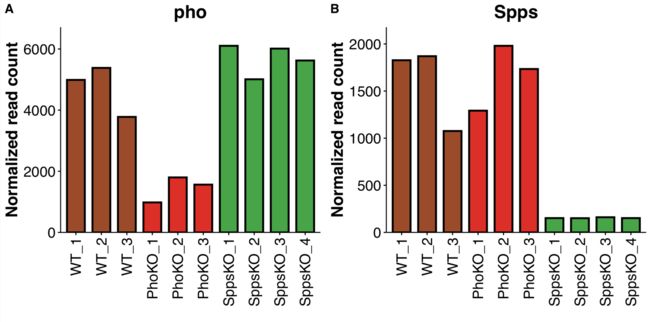
组合两个基因的图
Pho_plot <- my_barplot("pho")
Spps_plot <- my_barplot("Spps")
gene_exp_plot <- plot_grid(Pho_plot, Spps_plot,
labels = c("A", "B"))
# 保存到本地,由于我的电脑没有 `Arial` 字体,所以这里是报错的,故把 `family = "Arial"` 去掉
ggsave("gene_exp_plot.pdf", gene_exp_plot, width = 10, height = 5)
批量多个基因组图
gene_list <- a[, 1][1:5]
test <- lapply(gene_list, my_barplot)
# 每一行 最多五个图
five_gene <- plot_grid(plotlist = test, ncol = 5)
# 当然字体需自己调整一下了。
ggsave("five_gene_exp_plot.pdf", five_gene, width = 20, height = 5* length(test) %/% 5)

如果RNA-seq分析流程是自己走的,就会有bam文件或者bw文件,直接载入IGV也可以查看:
这一部分就跟着视频走了
链接: 生信 | IGV导出svg文件后期美化
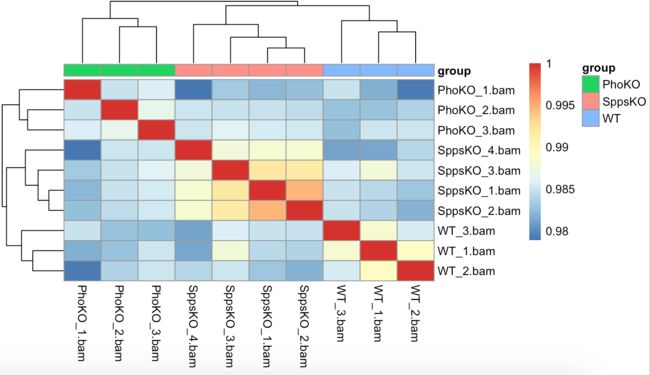
第二张图:相关性热图看不同样本相关性
当我们拿到数据时候,除了前面的质控等分析外,我们一般需要查看样品内的重复性怎么样,一般目前市面上的 RNA-seq、ChIP-seq 测序样品内的相关性都能高达 0.9 以上。
可以通过两种策略来计算样品内的相关性
1、根据基因的表达量信息来计算样品之间的相关性,比如 RNA-seq 。
2、将全基因组等分 bin 的方法,然后计算每个 bin 里面的 reads 数, 然后通过均一化等过程,再对数据进行计算相关性, 比如 ChIP-seq 等 DNA 类型测序数据。
热图一 通过基因的表达量来计算样品相关性
rm(list = ls())
options(stringsAsFactors = F)
a = read.table('~/Downloads/fly/RNA-seq/counts/all.counts.id.txt', header = T)
dim(a)
dat = a[,7:16]
library(stringr)
ac = data.frame(group=str_split(colnames(dat),'_',simplify = T)[,1])
rownames(ac) = colnames(dat)
M=cor(log(dat+1))
pheatmap::pheatmap(M,
annotation_col = ac,
# breaks = seq(0, 100, length.out = 100)
)
cov()函数计算相关性有三种方法:
Pearson,Kendall和Spearman三种相关分析方法参考 表观调控13张图之二相关性热图看不同样本相关性
热图二 分析deeptools软件的multiBigwigSummary和plotCorrelation得到的相关性结果
- linux/终端中运行:
multiBigwigSummary bins -b *.bw -o results.npz -p 4
plotCorrelation -in results.npz \
--corMethod spearman --skipZeros \
--plotTitle "Spearman Correlation of Read Counts" \
--whatToPlot heatmap --colorMap RdYlBu --plotNumbers \
--plotFileFormat pdf \
-o heatmap_SpearmanCorr_readCounts.pdf \
--outFileCorMatrix SpearmanCorr_readCounts.tab
- 将结果导入 R
b = read.table('SpearmanCorr_readCounts.tab', header = T)
colnames(b) <- str_split(colnames(b),'\\.',simplify = T)[, 1]
rownames(b) <- str_split(colnames(b),'\\.',simplify = T)[, 1]
bc = data.frame(group = str_split(colnames(b),'_',simplify = T)[, 1])
rownames(bc) <- colnames(b)
pheatmap::pheatmap(b,
annotation_col = bc)
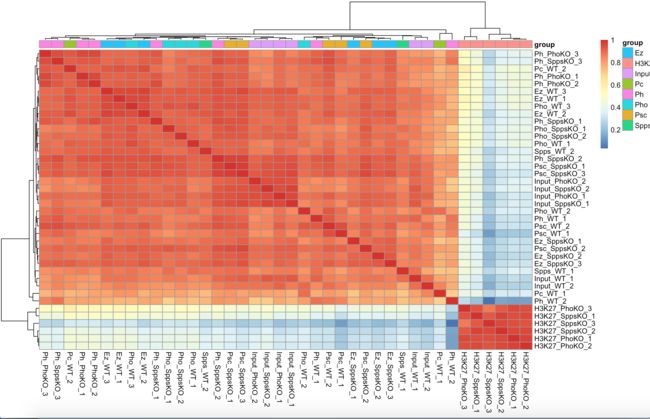
可以很清楚的看到,样品内的重复性都是极高的。同一样品都聚类在一起。样品内的相关性显著高于样品间的相关性。说明数据重复性很好,可以进行进下一步。(与前辈的有不一样,他展示了分析后的,我的就是原来是数据,没有进一分析。主要是关于这个)
第三张图 DESeq2寻找差异基因并可视化
DESeq2寻找差异基因
if(T){
# 赋值表达矩阵和分组信息为一个新的变量,方便以后直接调用
exprSet = dat
suppressMessages(library(DESeq2)) # 加载包
(colData <- data.frame(row.names = colnames(exprSet),
group_list = group_list) )
# 构建一个表达矩阵
dds <- DESeqDataSetFromMatrix(countData = exprSet,
colData = colData,
design = ~ group_list)
dds <- DESeq(dds)
# 下面我们得到 Spps 突变后的差异基因
res <- results(dds,
contrast=c("group_list", "SppsKO", "WT"))
# 注意这里是前面比后面,别把顺序搞反了,到时候一不注意结果就是反的。所以拿差异倍数对着原始 reads 比较一下。
resOrdered <- res[order(res$padj),] # 按照 padj 值进行排序
head(resOrdered)
DEG_SppsKO = as.data.frame(resOrdered) # 将数据转变为 data.frame 数据框
DEG_SppsKO = na.omit(DEG_SppsKO) # 去除包含 NA 值的行
# 下面我们得到 Pho 突变后的差异基因
table(group_list)
res <- results(dds,
contrast = c("group_list","PhoKO","WT"))
resOrdered <- res[order(res$padj),]
head(resOrdered)
DEG_PhoKO = as.data.frame(resOrdered)
DEG_PhoKO = na.omit(DEG_PhoKO)
# 将关键结果以 Rdata 形式保存到本地,下次如有需要就不需要重新用 DESeq2 计算了
save(DEG_PhoKO, DEG_SppsKO,file = 'deg_output.Rdata')
}
可视化
MAplot: 图 a 和 图 b
绘制 Spps
# apeglm 方法需要安装 apeglm 包
BiocManager::install("apeglm")
# ashr 方法同样需要安装额外依赖的包
BiocManager::install("ashr")
Spps_resLFC <- lfcShrink(dds, coef = "group_list_SppsKO_vs_PhoKO", type = "apeglm")
Spps_resNorm <- lfcShrink(dds, coef = "group_list_SppsKO_vs_PhoKO", type = "normal")
Spps_resAsh <- lfcShrink(dds, coef = "group_list_SppsKO_vs_PhoKO", type = "ashr")
par(mfrow=c(1,3), mar=c(4,4,2,1))
xlim <- c(1,1e5); ylim <- c(-3,3)
plotMA(Spps_resLFC, xlim = xlim, ylim = ylim, main = "apeglm")
plotMA(Spps_resNorm, xlim = xlim, ylim = ylim, main = "normal")
plotMA(Spps_resAsh, xlim = xlim, ylim = ylim, main = "ashr")
dev.off()
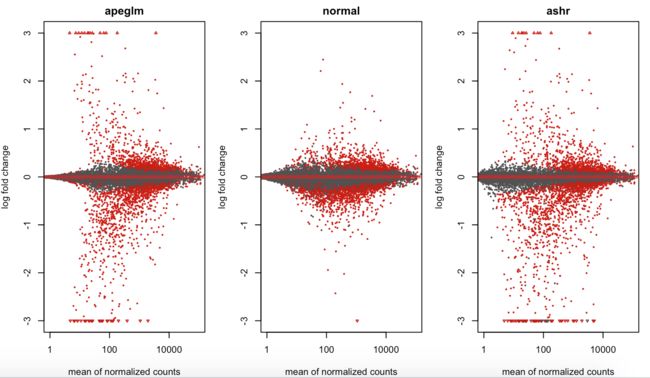
MAplot 在DESeq2 包中表示的是变化倍数 log2(Fold change) 与所有样本标准化后的 counts 数的平均值之间的关系。
怎么得到这个结果呢:
lfcShrink 收缩 FC 三种方法如下:
normalis the the originalDESeq2shrinkage estimator, an adaptive Normal distribution as prior. This is currently the default, although the default will likely change to apeglm in the October 2018 release given apeglm’s superior performance.apeglmis the adaptive t prior shrinkage estimator from theapeglmpackage (Zhu, Ibrahim, and Love 2018).ashris the adaptive shrinkage estimator from theashrpackage (Stephens 2016). HereDESeq2uses the ashr option to fit a mixture of Normal distributions to form the prior, with method="shrinkage".
这三种方法,后两种都是可以随便选择的。
同样的方法绘制另一个基因的图
Pho_resLFC <- lfcShrink(dds, coef = "group_list_WT_vs_PhoKO", type = "apeglm")
Pho_resNorm <- lfcShrink(dds, coef = "group_list_WT_vs_PhoKO", type = "normal")
Pho_resAsh <- lfcShrink(dds, coef = "group_list_WT_vs_PhoKO", type = "ashr")
par(mfrow=c(1,3), mar=c(4,4,2,1))
xlim <- c(1,1e5); ylim <- c(-3,3)
plotMA(Pho_resLFC, xlim = xlim, ylim = ylim, main = "apeglm")
plotMA(Pho_resNorm, xlim = xlim, ylim = ylim, main = "normal")
plotMA(Pho_resAsh, xlim = xlim, ylim = ylim, main = "ashr")
dev.off()
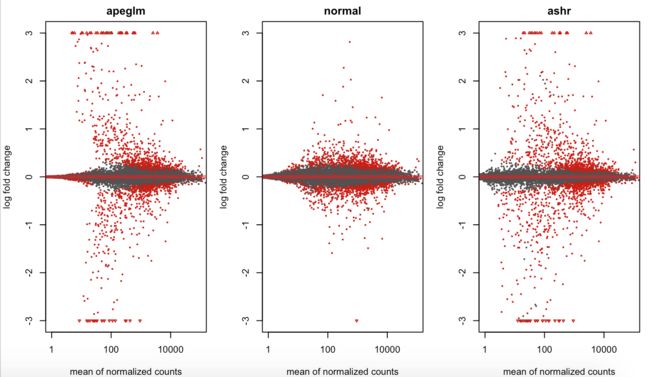
par(mfrow=c(1,2), mar=c(4,4,2,1))
plotMA(Spps_resAsh, xlim = xlim, ylim = ylim, main = "Spps_Ash")
plotMA(Pho_resAsh, xlim = xlim, ylim = ylim, main = "Pho_Ash")
dev.off()

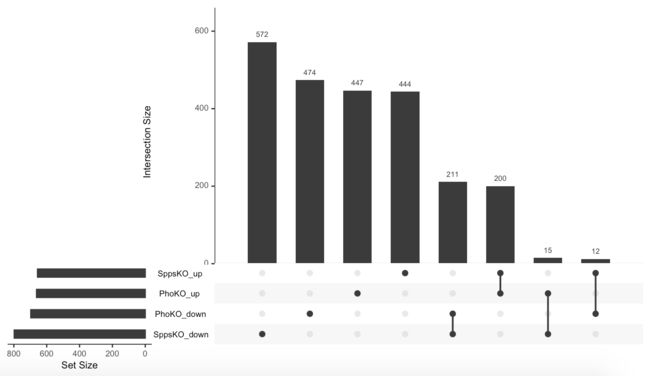
差异基因韦恩图:UpSetR/VennDiagram
第一种:我们使用 R 包 UpSetR 来绘制差异基因之间的韦恩图( 多组时候,这种更加一目了然 )
library(UpSetR)
load(file = 'deg_output.Rdata')
colnames(DEG_PhoKO)
DEG_PhoKO$change = ifelse(DEG_PhoKO$padj > 0.05,'stable',
ifelse(DEG_PhoKO$log2FoldChange > 0, 'up', 'down'))
DEG_SppsKO$change=ifelse(DEG_SppsKO$padj>0.05, 'stable',
ifelse(DEG_SppsKO$log2FoldChange > 0, 'up', 'down'))
SppsKO_up = rownames(DEG_SppsKO[DEG_SppsKO$change == 'up',])
SppsKO_down = rownames(DEG_SppsKO[DEG_SppsKO$change == 'down',])
PhoKO_up = rownames(DEG_PhoKO[DEG_PhoKO$change == 'up',])
PhoKO_down = rownames(DEG_PhoKO[DEG_PhoKO$change == 'down',])
allG = unique(c(SppsKO_up, SppsKO_down, PhoKO_up, PhoKO_down))
df = data.frame(allG = allG,
SppsKO_up = as.numeric(allG %in% SppsKO_up),
SppsKO_down = as.numeric(allG %in% SppsKO_down),
PhoKO_up = as.numeric(allG %in% PhoKO_up),
PhoKO_down = as.numeric(allG %in% PhoKO_down))
upset(df)
第二种:我们使用 VennDiagram来展示,也是就是文中那种花瓣图
# 这里直接 copy 琪音同学的
# 链接: https://mp.weixin.qq.com/s/Pg0mjz7mD73atMnHz7jv1A
# 导入本地字体,不然 `Arial` 字体会警告
#BiocManager::install("extrafont")
library("extrafont")
font_import()
loadfonts()
load(file = 'deg_output.Rdata')
colnames(DEG_PhoKO)
DEG_PhoKO$change = ifelse(DEG_PhoKO$padj > 0.05,'stable',
ifelse(DEG_PhoKO$log2FoldChange > 0, 'up', 'down'))
DEG_SppsKO$change=ifelse(DEG_SppsKO$padj>0.05, 'stable',
ifelse(DEG_SppsKO$log2FoldChange > 0, 'up', 'down'))
SppsKO_up = rownames(DEG_SppsKO[DEG_SppsKO$change == 'up',])
SppsKO_down = rownames(DEG_SppsKO[DEG_SppsKO$change == 'down',])
PhoKO_up = rownames(DEG_PhoKO[DEG_PhoKO$change == 'up',])
PhoKO_down = rownames(DEG_PhoKO[DEG_PhoKO$change == 'down',])
library(VennDiagram)
venn.diagram(
x = list(
"Up in phoKO" = PhoKO_up,
"Down in phoKO" = PhoKO_down,
"Up in SppsKO" = SppsKO_up,
"Down in SppsKO" = SppsKO_down
),
filename = "Venn.png", # 保存到当前目录
# stroke 颜色 形式 粗细
col = "#000000", lwd = 3, lty = 1,
# 填充 透明度
# 颜色可以参考前一篇,使用 takecolor 自己取色
fill = c("#D3E7F0", "#9FBEDB", "#A0D191", "#6DAE8A"),
alpha = 0.50,
# 里外字体大小形式
cex = 1.5,
fontfamily = "Arial",
fontface = "bold",
cat.cex = 1.5,
cat.dist = 0.2,
cat.fontfamily = "Arial",
# 图像整体位置 分辨率
margin = 0.2,
resolution = 300)

与文章趋势基本一致。可以看出 Spps 和 Pho 共同调控许多基因,说明这两基因有一定的关系。
两样本 log2FC 相关性散点图
load(file = 'deg_output.Rdata')
library(ggpubr)
DEG_PhoKO = DEG_PhoKO[rownames(DEG_SppsKO),]
po = data.frame(SppsKO = DEG_SppsKO$log2FoldChange,
PhoKO = DEG_PhoKO$log2FoldChange)
sp <- ggscatter(po, 'SppsKO', 'PhoKO',
add = "reg.line", # Add regressin line
add.params = list(color = "blue", fill = "lightgray"), # Customize reg. line
conf.int = TRUE # Add confidence interval
)
# Add correlation coefficient
sp + stat_cor(method = "pearson",
label.x = -10, label.y = 10) # 相关系数显示位置
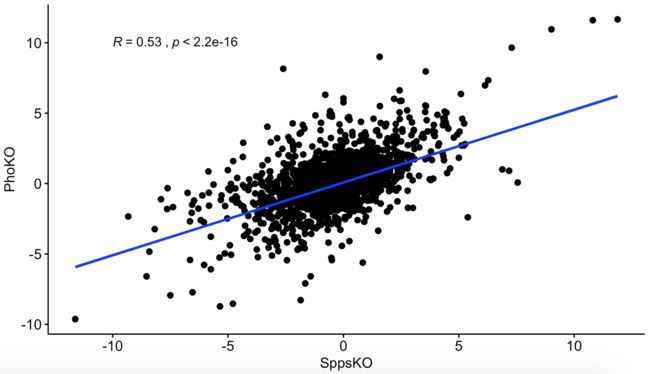
两者的相关系数高达0.53,这在两个基因间是算具有很强的相关的相关性了,更加佐证了上图的韦恩图的结果。
友情宣传:
- 广州专场(全年无休)GEO数据挖掘课,带你飞(1.11-1.12)
- 生信入门课全国巡讲2019收官--长沙站