第1篇
以一个基因为立足点的分析。发在EBiomedicine(6分)。
原文:High Expression of CPT1A Predicts Adverse Outcomes: A Potential Therapeutic Target for Acute Myeloid Leukemia
解读:如何从一个基因,整出两篇5分的文章
研究对象:CPT1A基因。
文章思路:
1、CPT1A基因在AML中的表达情况:相比正常人,白血病中显著高表达。
2、CPT1A基因与临床特征的关系:结果发现与年龄相关。
3、CPT1A基因与预后的关系:以CPT1A基因表达的中位数为界值分为高低表达分组,观察高表达和低表达组样本的预后差异,发现高表达组的预后更差。
4、CPT1A的功能:根据CPT1A高低表达组进行了差异分析,然后找到了差异表达的基因,进一步的利用这些基因做功能富集分析,找到了显著富集的通路,观察这些通路与白血病的关系,同时在差异基因中观察这些差异基因有没有被报道过与白血病相关,有相关的则列出来讨论。
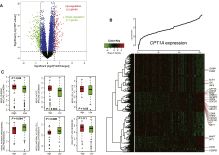
用的是差异分析的套路(分成两组,看差异基因):得到131 up-regulated and 271 down-regulated genes.
5、CPT1A与非编码RNA的关系:分析了高低表达组中miRNA的表达差异,并分析差异的miRNA与靶基因的关系,构建miRNA-mRNA网络,一顿分析差异的miRNA的功能,比如正相关microRNA包括miR-222,miR-221,miR-20a,miR-17,miR-155,miR-26a,miR-335等等。已发现所有这些microRNA在先前的研究中具有重要的肿瘤促进价值。如miR-222 / 221可以增强黑素瘤细胞的增殖和分化阻断等。

这个火山图是p值与系数的二维图(不是FC值),因此不是分为CPT1A高低表达组,而是以连续变量的形式分析。另外这个热图,并不是简单的聚类图,横坐标是CPT1A表达从低到高排列(只对特征进行聚类,而不对样本聚类,样本直接根据CPT1A表达排序)。
这个 miRNA-mRNA网络有点意思,可以学习下:To further clarify the biological functions of microRNAs, interaction-network was constructed based on the overlapping of results derived from microRNA-target predicting algorithm ([Garcia et al., 2011](javascript:void(0);)) and correlation analysis.
6、CPT1A高低表达与甲基化的关系:与甲基化的关系主要有两方面,甲基转移酶的表达和全基因组甲基化水平;首先我们分析CPT1A高低表达组中甲基转移酶的表达差异,发现他们在高CPT1A组中表达高;进一步的我们分析CPT1A高低表达组中全基因组甲基化的差异(简单来说就是CPT1A高低表达组中的甲基化差异分析),得到差异的甲基化位点,根据这些位点的基因组位置,统计一下他们在启动子区域是怎样的一种分布形式以及在CpG岛上市怎样的一种分布形式(甲基化分析常规套路)。

也是用的差异表达分析的套路。
关于甲基转移酶的知识: DNA 甲基转移酶的表达调控及主要生物学功能
第2篇
以一个基因为立足点的分析。发在Front. Immunol(5分)。分析套路很一般。
原文:LAYN Is a Prognostic Biomarker and Correlated With Immune Infiltrates in Gastric and Colon Cancers
解读:如何从一个基因,整出两篇5分的文章
这是一篇发表在五分多的杂志上关于LAYN是作为一种预后生物标志物与胃癌和结肠癌中的免疫浸润相关的分析的文章
1、首先利用Oncomine数据库分析LAYN基因在各个肿瘤中的表达差异情况,发现在乳腺癌,结肠直肠癌,胃癌,肾癌,胰腺癌和淋巴瘤肿瘤中的LAYN表达更高,在膀胱癌,乳腺癌,结肠直肠癌,头颈癌,肺癌,卵巢癌和前列腺癌中观察到较低的表达,进一步的利用TCGA数据库的RNAseq数据分析验证了一遍,结果似乎有点点类似。

2、LAYN基因与预后的关系:利用PrognoScan、GEPIA等在线工具(两个不用写代码的工具)分别分析了LAYN在各个肿瘤中的预后差异,发现LAYN表达显着影响5种癌症的预后,包括结肠直肠癌,乳腺癌,眼癌、卵巢癌和胃癌
3、胃癌样本中LAYN基因表达与临床特征的关系,发现了一些显著性(个人觉得没太大意思)。
4、肿瘤浸润性淋巴细胞是在癌症前哨淋巴结状态和存活的独立预测因子,研究了LAYN表达是否与不同类型癌症中的免疫浸润水平相关,首先我们从TIMER数据库(Cibersort类似功能)下载了39种癌症的六种免疫浸润细胞的得分数据,分别分析了LAYN表达与这些免疫细胞得分的相关性,发现在CODA和STAD中,LAYN表达水平与CD8+ T cells 、CD4+ T cells、macrophages 、neutrophils和DCs 有显著正相关。这个结果还可以。

5、既然在CODA和STAD中LAYN表达于免疫细胞这么相关,便找来一些其他已报到的免疫浸润相关的基因集,看看这些基因集与LAYN的相关性,结果显示LAYN表达水平与各种免疫细胞和COAD和STAD中不同T细胞的大多数免疫标记物组显着相关。
第3篇
解读:经典套路解析:胃癌亚型预后分析(CCR,IF=10.19)
直接看上述的解读吧,不再赘述。经典的套路了。
第4篇
免疫相关的经典文献。很值得研读的一篇,里面涉及了很多经典的方法工具。
原文:Identification of genetic determinants of breast cancer immune phenotypes by integrative genome-scale analysis
解读:一篇文献一个技术路线:火热的免疫
在上述解读的基础上进行一些补充吧。
1、关于一致性聚类,除了文献中提到的“根据Calinski指数确定最佳聚类数”,还可以使用PAC的方法确定聚类数,灵活使用,根据自己的需求。
2、免疫细胞组成的刻画。这里没有使用CiberSort系列方法,很奇怪。这里用ssGSEA的方法(GSVA包里集成了),为每个样本计算各个免疫细胞的ES分数。
3、免疫表型间差异基因的功能富集分析。 ICR4 vs ICR1的差异基因分析DEG,这里是常规的方法,然后对差异基因进行GSEA的功能富集,再可视化即可。这一步和上一步似乎没有连续性,只是从不同角度刻画了这些信息。
4、MAPK突变得分(MAPK-mut score)。将Luminal型人群根据有无MAP3K1/MAP2K4突变分成2组,在MAPK通路中基因进行差异分析(KEGG可获得该通路的基因),从而计算MAPK-mut score,用来进一步区分Luminal型人群。
总结:本文分析的角度比较多,但没有一条很明确的主线,有点像方法的堆积,花里胡哨。但里面的各种分析方法也值得学习:比如新抗原、突变负荷、各免疫细胞得分、驱动基因等。
第5篇
深度学习与多组学(虽然个人觉得这个模型只能叫浅层神经网络)
原文:Deep Learning–Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer
解读:一文献一技术路线:人工智能+肿瘤预后
没有特别的生信套路,主要思路是利用DL模型建立预后模型,然后和传统方法进行对比,发现优势明显。再就是,考虑组学数据和临床数据的结合,发现临床数据再加入没有必要(C-index没有上升)。
具备生信特色的是,也进行了差异分析和功能富集分析。这个逻辑有点怪:差异分析的分组是预后模型得到的分组(高危与低危),也就是说寻找高低危组的差异基因,然后看看功能如何。奇怪之处在于,这个高低危分组源于多组学测序数据(包含了RNA-Seq),又回过头来找RNA-Seq中的差异基因。
值得借鉴的是,之后遇到多组学的数据(甚至就是单组学数据),也可以尝试神经网络建模的方法(可惜不是真正的深度模型啊)。
第6篇
泛癌分析,讲端粒酶特征基因与预后关系的
原文:Pan-cancer analysis identifies telomerase-associated signatures and cancer subtypes
解读:不是癌的cancer:泛癌(Pan-Cancer)
端粒酶相关基因列表来自先前别人的研究报道。
结合了多组学数据:转录、突变及CNA、miRNA、甲基化。
这里使用了PSM倾向性评分,发挥得好像不错,使用了权重的概念,并且构建了个优化FC的东西。
miRNA与mRNA互作,使用了miRtarBase,miRanda和miRDB数据库。通过WGCNA识别miRNA-mRNA共表达模块。
药物靶标分析(留个心眼,以后也可以试试)。
关于hub基因:In the gene coexpression module network, hub genes with high intramodular connectivity represent a small proportion of nodes with maximal information compared to other nodes. Hub genes play important roles in the module network.
第7篇
原文:Novel RB1-Loss Transcriptomic Signature Is Associated with Poor Clinical Outcomes across Cancer Types
解读:一文献一思路:“泛癌”是什么癌?
这篇文献的解读补充如下
1-关于186基因的筛选构建:采用PAM方法,可以收缩筛选基因(类似于LASSO的作用)。再利用后验输出概率,可以得到分类得分RBS(类似于Logistic的输出概率)。这一套似乎可以用LASSO-Logistic替代,大部分场景下 孰优孰劣?
2-评估186基因谱的usefulness,采用了t-SNE的方法。t-SNA两大主成分看到是能分开的,疑问:为啥不使用PCA或者其他主成分分析?
3-评估甲基化与RBS的关系。这个有点意思的。其实就是依赖于一个观点:基因启动区上游的高甲基化导致基因的低表达。这里RBS与甲基化的相关性,与这个观点相符。再次提供了RBS有效的证据(多个层次说明模型的有效性)
4-对RBS评分再次分类,并进行生存分析。个人不理解的是:为什么不直接使用PAM的分类结果(阈值为0.5),而要使用Youden Index法重新寻找界值0.6?生存分析效果不好?
文章展示了常规拷贝数变化的生存分析和RBS定义丢失的生存分析KM曲线。
5-在转移性去势抵抗前列腺癌队列(mCRPC)中验证说明了2个问题。第一个是:RNA-Seq(RBS)与DNA-Seq(拷贝数变化)的一致性很高(AUC达到0.9),这个在其他癌症里没这么明显。第二个问题是,RNA-Seq(RBS)与DNA-Seq(拷贝数变化)在多因素分析里都是显著的,具有独立预后作用,由此说明RBS可以作为预测预后的补充。作者想间接说明:RBS可以探测到DNA测序无法评估的RB1功能丢失的情况。原文:
We found that both the RNA-seq and DNA-sequencing definitions were independently predictive of short OS (P = 0.0036 and P = 0.046, respectively), suggesting that both RNA-seq and DNA sequencing offered unique information on RB1 status that could be used to detect a clinical phenotype of RB1-impaired, clinically aggressive mCRPC.
这个思路挺有意思的。
第8篇
原文:Integrative Analysis Defines Distinct Prognostic Subgroups of Intrahepatic Cholangiocarcinoma
解读:一文一路线:“粗暴”的多组学
解读的补充:
1-两个R工具:MeDeCom包,可以通过DNA甲基化谱刻画其相应细胞组成情况;LUMP方法可以通过甲基化数据计算肿瘤纯度(肿瘤纯度估算的4大方法之一)。
2-拷贝数变异分析工具:GISTIC,用于分析拷贝数变异的工具。用于在肿瘤的众多SCNA中选择出具有统计学意义的SCNA,排除背景率的干扰。
3-甲基化的位置富集分析,使用18‐state ChromHMM model of Roadmap Epigenomics。如图
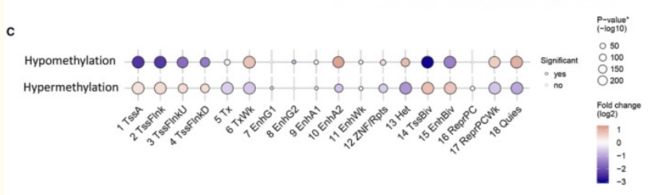
甲基化的转录因子结合位点富集分析,使用 transcription factor binding sites from ENCODE。如图

甲基化的功能富集分析。类似于转录组的功能富集,使用GREAT tool and the GO Molecular Function database。
4-多组学聚类分析:iClusterPlus。学习一个。
这篇文章没完全嚼透,之后有时间再深入解读。
第9篇
原文:Survival Analysis of Multi-Omics Data Identifies Potential Prognostic Markers of Pancreatic Ductal Adenocarcinoma
解读:“多组学验证”高分标配
解读补充:
相关性图的花哨做法:

这篇文献整体来看没啥意思。分析虽广但不深刻,方法比较平淡。
第10篇
原文:Genomic Signatures Predict the Immunogenicity of BRCA-Deficient Breast Cancer
解读:解决临床问题,这是科研应有的样子
解读补充:
1-关于cytolytic index的概念。这个东西以前没听过,好像翻译成“溶细胞”的能力?与免疫细胞发挥杀伤作用有关?以后免疫分析时可以尝试下这个。文中这么写的:
Cytolytic index for each tumor was computed as the geometric mean of PRF1 and GZMA expression from RNAseq v2 expression data downloaded from cBioportal
顺带的,这篇关于cytolytic的文献也可以看看:Immune Cytolytic Activity Stratifies Molecular Subsets of Human Pancreatic Cancer
。还有一篇2015年的Cell:Molecular and Genetic Properties of Tumors Associated with Local Immune Cytolytic Activity。
2-从多个变量的角度来研究,建立层级分类树。
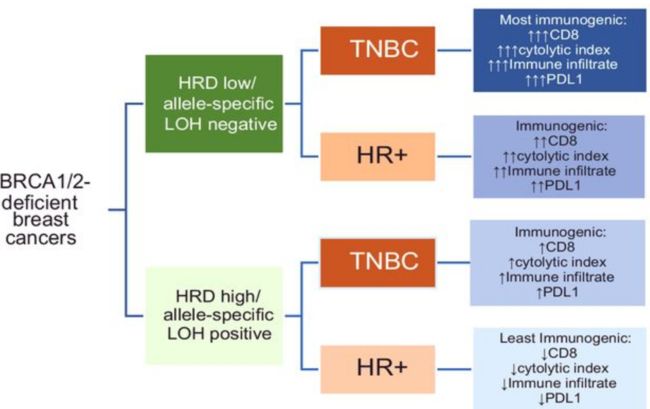
这种做法是值得学习的。看上去纵向思路并不深,但是却以横向思路整合成纵向。
之后做多各潜在指标进行刻画时,可以采取这样的策略。另一方面,也可以用这几个指标建模。都可以尝试。
注:文中或图中的HR+等价于Rec+,指的是激素受体阳性,与HR(同源重组)无关。而HR与HRD的概念,文中也是重点指标;HRD是计算出的HR缺失得分(计算方法略,文中方法部分有提到,是直接使用了一个R-script和Sequenza),与HR是否阳性是不同的。
3-新抗原负荷。似乎这个新抗原负荷及肿瘤负荷,与免疫浸润不呈正比(文中的图表),可能我理解错了。新抗原负荷这个指标要怎么去使用呢?以哦户可以思考下。
4-GSVA这个工具,看来我还是不够熟练啊,之前就简单得当成了GSEA的替代品。得再研究下。
这篇文献值得学习的,Marker一个。