果蝇探索PRC复合物之文章复现(一)
老大曾在公众号上发过一篇推文:在果蝇探索PRC复合物(逆向收费读文献2019-18),里面提到了
表观领域的一篇文章:Global changes of H3K27me3 domains and Polycomb group protein distribution in the absence of recruiters Spps or Pho。这篇文章据说是RNA-seq和ChIP-seq数据分析结合的典范。关于PRC复合物,文章中的结论是-PcG recruiters, the PRC2 component E(z), and the PRC1 components Psc and Ph cobind thousands of active genes outside of H3K27me3 domains.关于如何获取数据也就是拿到peaks文件和表达矩阵,可以去哔哩哔哩网站搜索生信技能树Chip—seq测序数据分析视频。其实呢老大已经录制好了关于这篇文章中图表复现的视频,我这里跟着视频,复现文章中的图图。
- 第一个复现的图-Figure S12 Decreased expression of Pho and Spps in the corresponding mutants
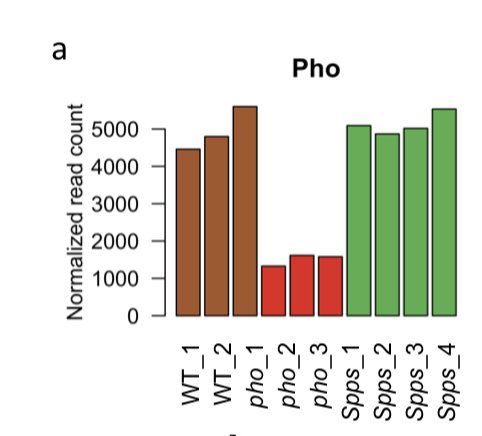
上图来自做完RNA-seq后的counts值,如果还不会linux的小伙伴又想直接练习R中的代码,可以问我要counts矩阵。
先要读取表达矩阵,表达矩阵来自RNA-seq的feature counts的结果。也就是文件all.counts.id.txt。
读进来的表达矩阵如下图
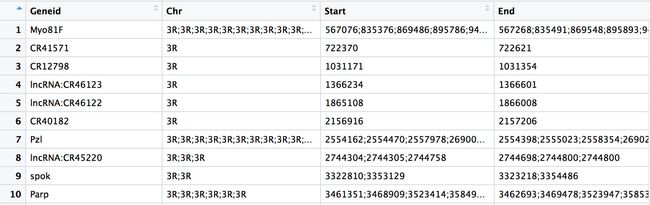
- 第一列:基因名
- 第二列:染色体
- 第三列:基因起始坐标
- 第四列:基因终止坐标
- 第五列:略
为什么有些基因有这么多的其实坐标和终止坐标呢?是因为有些基因有多个外显子。
在复现上面的第一张图片前,老大出题儿让我对这个表达矩阵做进一步了解。
题目一
挑选出基因名前两位为AB或者ab的基因名,并挑选出长度最长的基因
ab <- a[grep('^AB',a$Geneid,ignore.case = T),]

lapply(1:nrow(ab),function(i){
sum(as.numeric(strsplit(ab[i,4],';')[[1]])-as.numeric(strsplit(ab[i,3],';')[[1]]))
})

which.max(lapply(1:nrow(ab),function(i){
sum(as.numeric(strsplit(ab[i,4],';')[[1]])-as.numeric(strsplit(ab[i,3],';')[[1]]))
}))

ab[which.max(lapply(1:nrow(ab),function(i){
sum(as.numeric(strsplit(ab[i,4],';')[[1]])-as.numeric(strsplit(ab[i,3],';')[[1]]))
})),1]

或者可以
ab <- a[grep('^AB',a$Geneid,ignore.case = T),]
tmp2 <- apply(ab,1, function(i){
sum(as.numeric(strsplit(i[4],';')[[1]])-as.numeric(strsplit(i[3],';')[[1]]))
})

names(tmp) <- ab$Geneid#这一步是给向量tmp加上基因名,用names函数,给向量加基因名用names函数
tmp

names(which.max(tmp))
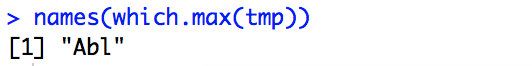
题目二:chr这一列有多少种元素

tmp3 <- as.data.frame(a$Chr)
unique(apply(tmp3,1, function(i){
unique(sort(str_split(i,';',simplify = T)[1,]))
}))
上面的代码有点长,需要分解一下
apply(tmp3,1, function(i){
unique(sort(str_split(i,';',simplify = T)[1,]))
})
得到的结果有17714行,实际上这个a矩阵就是有这么多。那么我们在用uniq去重复就好了

现在对第一张图复现,图是敲了Pho后看看敲除后的效果,rna-seq的表达量是否降低。
rm(list = ls())
options(stringsAsFactors = F)
a=read.table('all.counts.id.txt',header = T)
dim(a)
cg1=a[a[,1]=='pho',7:16]
library(ggpubr)
library(stringr)
dat=data.frame(gene=as.numeric(cg),
sample=names(cg),
group=str_split(names(cg),'_',simplify = T)[,1]
)
ggbarplot(dat,x='sample',y='gene',color = 'group')
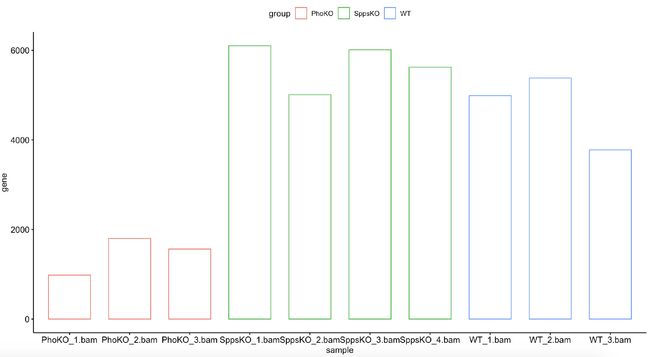
可以看到虽然上面的图和文章的图图还有些差别,关于作图可以再另一个作图的板块细细的写一下!上图可以看出Pho敲除组的表达量是明显低于其他组的,就OK了。
上面的代码均来自生信技能树哦
最后友情宣传生信技能树
生物信息学“义诊”
生物信息学"拍卖会"
全国巡讲:R基础,Linux基础和RNA-seq实战演练 : 预告:12月28-30长沙站
广州珠江新城GEO数据挖掘滚动开班
DNA及RNA甲基化数据分析与课题设计