NGSQC Toolkit 用于进行高通量测序数据的质量检查和数据过滤,具有去接头,去除非ATGC碱基,去除低质量碱基和reads等功能
1 安装
下载NGSQC Toolkit http://www.nipgr.res.in/ngsqctoolkit.html

在linux下解压缩,并给予执行权限
>unzip NGSQCToolkit_v2.3.3.zip
>find/data/home/chjiao/software/NGSQCToolkit_v2.3.3 -name '*.pl' -exec chmod 755 {} \;
2 安装libgd和GD模块
cpan
cpan>installString::Approx
cpan>GD
cpan>GD::Graph #需要安装libgd后才能正确安装
3 NGSQC Toolkit介绍
NGSQC Toolkit包解压后产生4个文件夹,行使以下功能:去接头、去除低质量的碱基、去除模糊的碱基N、格式转换、统计。
QC文件夹含有4个perl程序,能去除454或者illumina reads的adaptor或者低质量的reads。
因为我的数据是illumina测的,所以以IlluQC.pl为例,讲一讲具体使用。
3.1 IlluQC.pl
用于Illumina reads的QC。输入文件为fastq或者fastq的任何变化形式,默认情况下去除掉含有primer/adaptor的reads和低质量的reads,并给出统计结果和6种图形结果。
使用:perl IlluQC.pl
必需参数:
双端:
-pe
eg: -pe r1.fq r2.fq 31
单端:
-se
单端与双端的参数只在输入文件上有区别,双端输入两个文件,单端输入一个文件
eg:-se r1.fq 3 2
接头库的选择
1 = GenomicDNA/Chip-seq Library
2 = Paired End DNALibrary
3 = DpnII geneexpression Library
4 = NlaIII geneexpression Library
5 = Small RNALibrary
6 = Multiplexing DNALibrary
按照自己测序数据来选择,因为我的数据是RNA双端测序数据,这里的接头库我选择2
FASTQ 格式的选择(不确定的话,用A选项就好,系统会自动的检测fastq的格式)
1 = Sanger(Phred+33, 33 to 73)
2 = Solexa(Phred+64, 59 to 104)
3 = Illumina (1.3+)(Phred+64, 64 to 104)
4 = Illumina (1.5+)(Phred+64, 66 to 104)
5 = Illumina (1.8+)(Phred+33, 33 to 74)
A = Automaticdetection of FASTQ variant
其他参数:
-l设定质量的read长度的百分比的截断值(0-100)默认是70,也就是低质量碱基在reads中比例 >30% 的为低质量reads
-s 过滤碱基质量截断值(整数0-40),默认是20,也就是说碱基质量低于20的即为低质量碱基
-p 线程
-t 输出统计结果的格式,默认是1
1=formatted text
2=tab delimited
-o 输出路径
-z 输出文件的压缩格式,默认是t
t = text FASTQfiles
g = gzipcompressed files
3.2 IlluQC_PRLL.pl
IlluQC_PRLL.pl与IlluQC.pl程序的差别仅仅是多了一个参数
-c 设置cpu,提高运行速度
3.3 AmbiguityFiltering.pl
去除reads中的非ATGC碱基
使用:perl ..\AmbiguityFiltering.pl
必需参数:
-i 输入文件(fastq/fasta格式)
-irev 输入文件(如果是双端测序,这里输入双端测序的第二个文件,可选)
eg:-i f1.fq -irev f2.fq
其他参数:
-c 允许的非ATGC碱基的最大数量默认0
-p 允许的非ATGC碱基的最大比例默认0
-t5 从5'端修剪非ATGC碱基
-t3 从3'端修剪非ATGC碱基
注意:以上4中修建模式4选1
-n 设置丢弃的reads长度,当reads长度低于设定值时会被丢弃默认-1
-o 输出路径
3.4 TrimmingReads.pl
去除低质量的碱基和reads
使用: perl..\TrimmingReads.pl
必需参数:
-i 输入文件(fastq/fasta格式)
-irev 输入文件(如果是双端测序,这里输入双端测序的第二个文件,可选)
eg:-i f1.fq -irev f2.fq
其他参数:
-l 从5'端修剪的碱基数
-r 从3'端修剪的碱基数
-q 从3'端修剪质量分数低于设定阈值的碱基,默认为0
注意:只有当-l或者-r不执行的时候,-q才能设置
-n 设置丢弃的reads长度,当reads长度低于设定值时会被丢弃默认-1
-o 输出路径
3.5 统计工具
NGSToolki提供了AvgQuality.pl N50Stat.pl两个程序来统计数据的平均质量和N50,因为我用NGSToolki处理完会直接用fastqc来查看数据质量,所以这里就不累述这两个程序的功能。
3.6 格式转换工具
NGSToolki提供了
SangerFastqToIlluFastq.pl
SolexaFastqToIlluFastq.pl
FastqTo454.pl
FastqToFasta.pl
4个程序可以将Sanger测序、Illumina测序、454测序、fastq和fasta格式进行转换。
也不累述了,可直接看官方文档。
4 实战:
数据质量问题:
1 测序质量整体偏低,特别是73个碱基后的序列质量,是由于73个碱基后的序列非ATGC碱基的含量很多
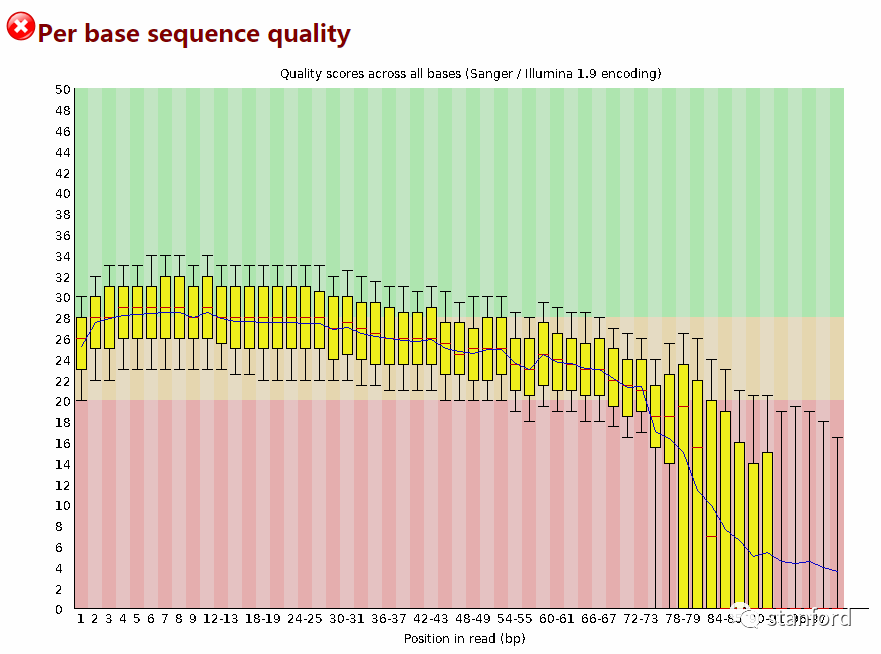
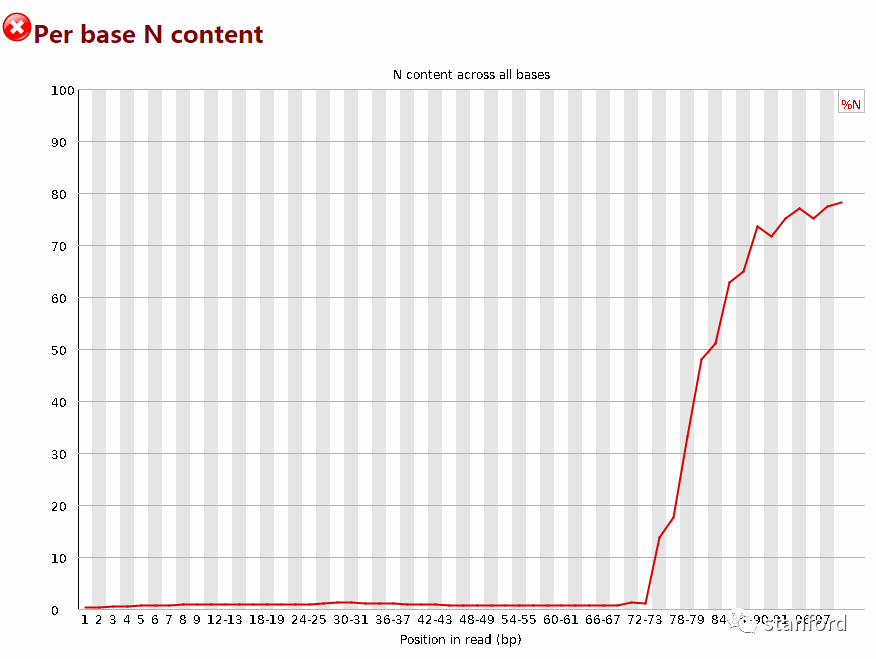
2 少量的接头
3 ATGC的含量在reads的后半段依旧是不均衡的


NGSQC Toolkit过滤策略:
1 利用IlluQC_PRLL.pl程序去除接头和质量低于20的碱基以及低质量的reads。
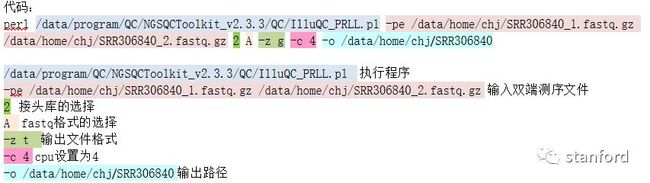
输出结果为:
2个过滤后的fastq文件,和一个html报告文件
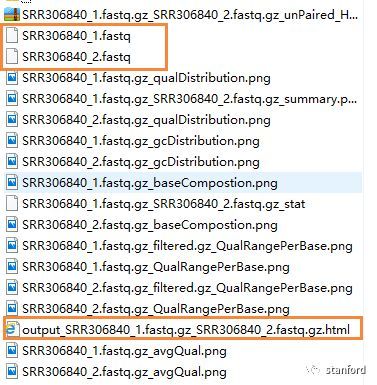
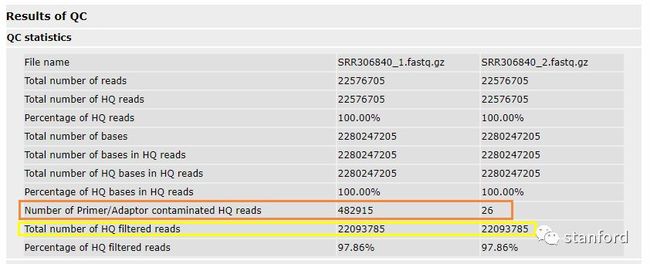
报告文件包含一些基本的统计信息,包括接头含量,低质量的reads含量,过滤完接头和低质量的reads之后的reads含量等信息。这里我们可以看出我们高质量的reads的比例是100%,去完街头后的reads数是22093785.
2 用AmbiguityFiltering.pl程序去除reads中的非ATGC碱基N

输出结果:2个过滤后的fastq文件

3 去除低质量的reads

输出结果:2个过滤后的fastq文件
4 用fastq再次检测过滤后的fastq文件的质量

过滤前后数据质量变化如下:
1 Total Sequences
过滤前:22576705
过滤后:3832289
因为我们设置的非ATGC碱基的含量为0,条件比较苛刻,所以过滤掉了大量的reads。
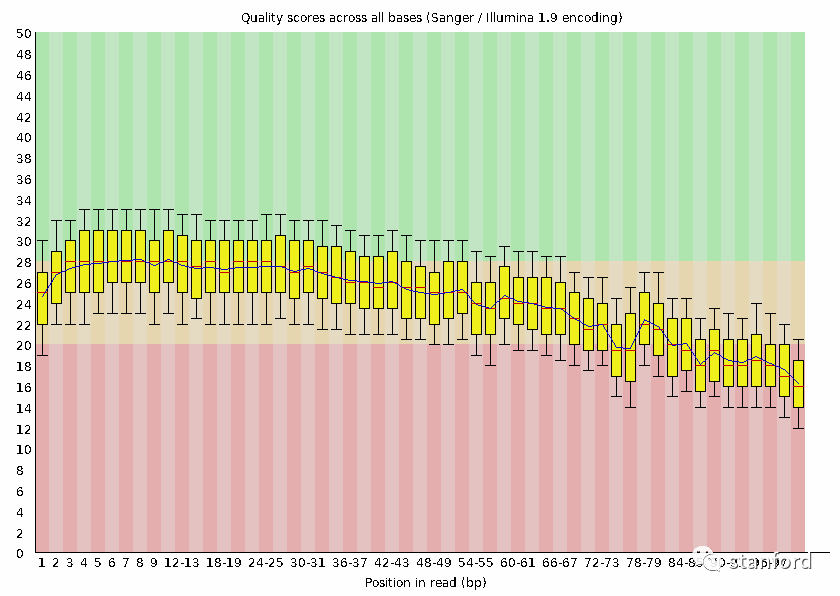
2 测序质量:在reads后半段的平均碱基质量有大幅度的提升
过滤前:

过滤后:
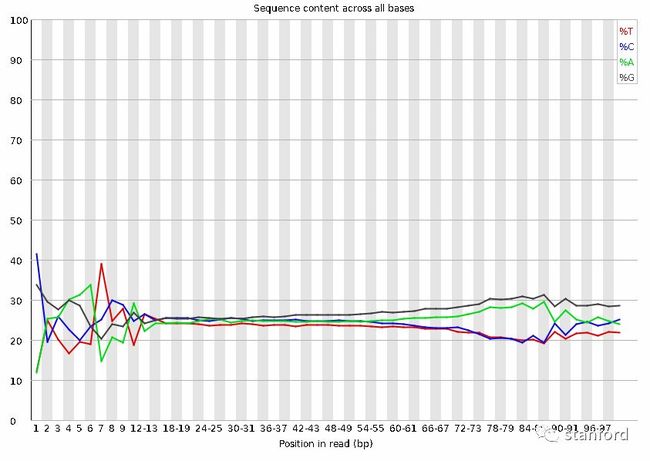
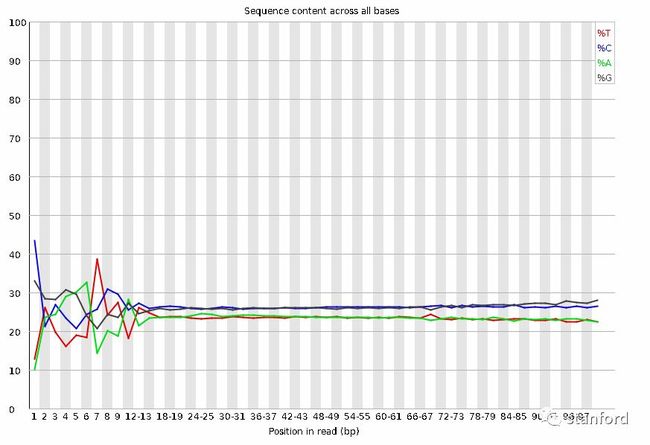
3 Per base sequence content
统计每个位点的ATGC含量,reads后半段的ATGC含量曲线趋近于平行
过滤前:
过滤后:
4 非ATGC碱基含量
清除了所有的非ATGC碱基,非ATGC碱基曲线为百分之0
过滤前:

过滤后:
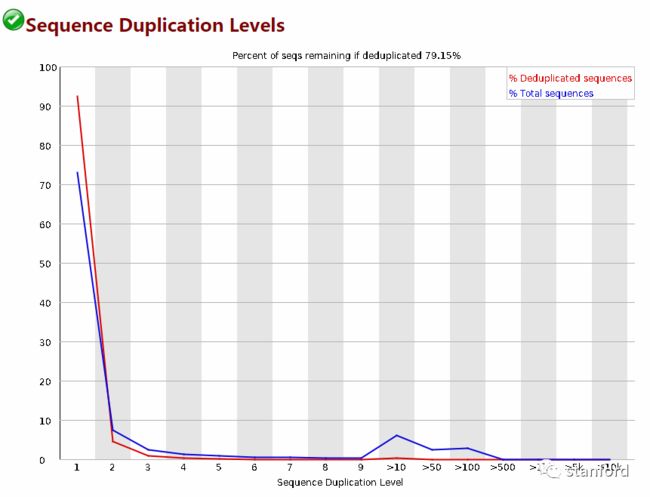
5 duplicationreads 的比例:
当duplication reads比例>20%时,报warning。过滤后duplicationreads比例低于20%
过滤前:

过滤后:
6 Overrepresented sequences过表达序列:
过滤后无过表达序列
过滤前:

过滤后:

7 接头:接头含量降低
过滤前:
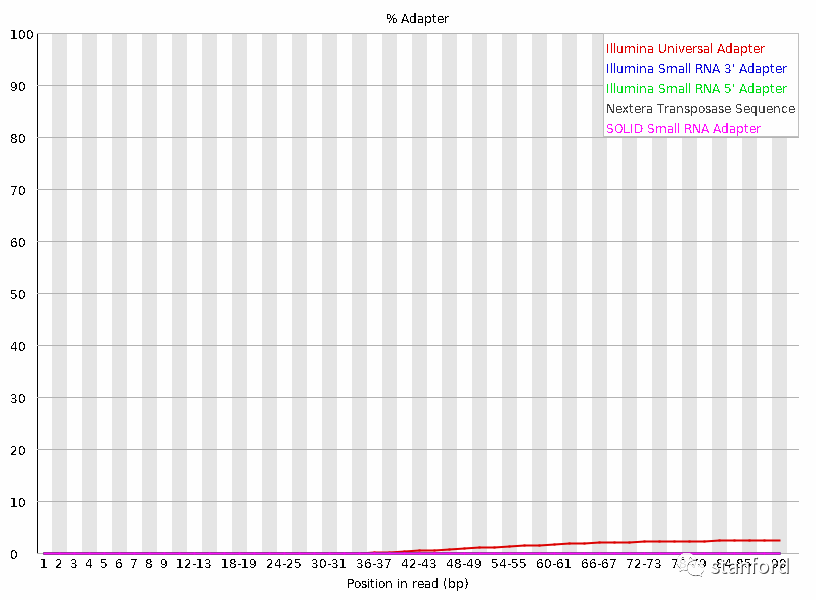
过滤后:

问题:
1根据每个位置的碱基平均质量图,可以看出有些碱基的质量低于20,但在用NGSQC Toolkit的过滤低质量碱基的参数,-q 20(从3'端修剪质量分数低于20的碱基)且默认设置当低质量碱基高于30%时则认为是低质量reads,过滤后结果显示我的高质量reads是100%,未过滤任何低质量的reads。
可能原因:reads中低质量碱基的比例不超过30%,则被判定为高质量reads。
解决办法:后期想利用另一个软件Trimmomatic来试试去除低质量的reads
2 数据GC含量为51%,人类的基因组GC含量在40%左右,后期利用去污染软件kraken,去除其他物种的污染。
吐槽:数据质量太差了,坑好多哇,哭唧唧~另外生信小白痴一枚,要是有大神看不下去给我辅导辅导也是极好的~
