最近开始打算学习一下基本转录组分析,在此做一下笔记记录遇到的各种坑。
文章用的是NC的一片人类转录组的文https://www.nature.com/articles/ncomms13347。
1、数据准备
在文章中可以轻易找到GEO数据的accession ID GSE81916。然后去NCBI的GEO数据库下搜索下载该实验的所有数据。
1.1 使用 fastq-dump命令从sra文件中提取fq文件
sra数据下载完后使用sratoolkit里面的fastq-dump --split-3 SRR* 来将sra转换为相应的fq文件,命令如下:
$ for file in `ls *.sra`; do (nohup fastq-dump --gzip --split-3 ${file} -O rawdata/ 1>fq_dump.log 2>&1 &) ;done
若SRA文件中只有一个文件,那么--split-3这个参数就会被忽略。如果原文件中有两个文件,那么它就会把成对的文件按*_1.fastq, *_2.fastq这样分开,分别是左右两端测得的序列。如果还有出现了第三个文件,就意味着这个文件本身是未成配对的部分。可能是当初提交的时候因为事先过滤过了一下,所以有一部分数据被删除了。
fastq-dump --split-files SRR*结果类似fastq-dump --split-3,它仅生成两个配对的文件_1.fastq和_2.fastq,而忽略了未配对的文件。
1.2 记录一下下载数据时遇到的坑
在得到所有RUN的id后,用两种简单的方式可以下载:NCBI提供的工具sratoolkit里面的prefetch或者aspera connect。这两种方式都可以在我的另一篇笔记里找到安装及使用方法。
在这里我选择使用的是aspera connect, 因为他可以方便的选择数据下载在哪里。但是在写批量下载的时候出现了问题,一直下载不了,后来发现是shell脚本里面无法使用自己命名的命令,如alias的这种都不行,坑了个吧钟头。最后使用的下载数据的命令如下:
#aspera connect 下载的脚本
#!/bin/bash
# Usage: asp SRRID download_dir
SRA=$1
DIR=$2
ascp -v -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh -k 1 -QTr -l 200m [email protected]:/sra/sra-instant/reads/ByRun/sra/SRR/${SRA:0:6}/${SRA}/${SRA}.sra ${DIR}
然后给ascp alias一下方便使用:
echo alias asp="/home/sxuan/scripts/ascp.sh" > ~/.bashrc
. .bashrc
# 之后便可以直接使用下面命令下载
asp SRR2854733 download_dir
最后下载数据使用下面的命令批量下载数据:
nohup cat SRR_Acc_List.txt| while read id; do asp $id . ; done > download.log 2>&1 &
当时就是把这一行命令写进脚本里面去执行发现不行,坑了大半天才反应过来。
2、数据质控
从sra文件中提取得到fq数据后,使用fastqc查看数据质量如何,命令如下:
for j in `seq 1 2`; do for i in `seq 56 62`; do (nohup fastqc -t 5 -O qc "SRR35899"${i}"_"${j}".fastq.gz" 1>>qc.log 2>&1 &);done;done
查看结果发现,序列的前几个碱基很不稳定,如下图:
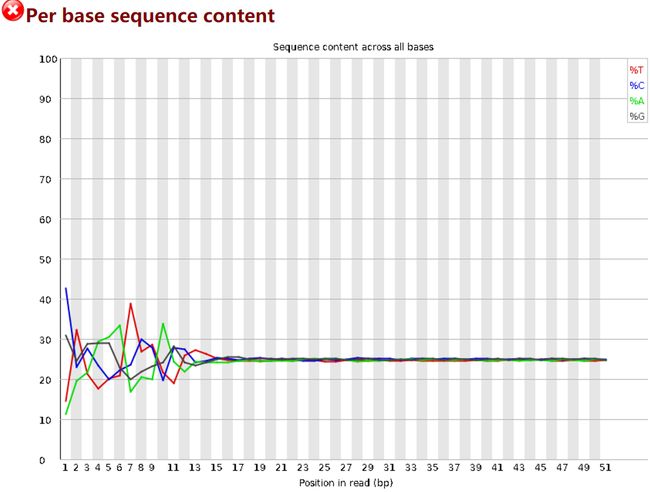
因此使用
trimmomatic去除所有文件的每条序列的前10个bp,保留长度大于20bp的序列,命令如下:
#!/bin/bash
for i in `seq 56 62`;do
trimmomatic PE -threads 4 -phred33 ~/RNA/rawdata/SRR35899${i}_1.fastq.gz ~/RNA/rawdata/SRR35899${i}_2.fastq.gz ~/RNA/cleandata/SRR35899${i}_1_paired.fastq.gz ~/RNA/cleandata/SRR35899${i}_1_unpaired.fastq.gz ~/RNA/cleandata/SRR35899${i}_2_paired.fastq.gz ~/RNA/cleandata/SRR35899${i}_2_unpaired.fastq.gz HEADCROP:10 MINLEN:20
done
3、Mapping
3.1 建立Index
数据处理完之后便可以开始进行比对了。但在正式比对之前还需要先对参考基因组建立索引,也可以直接去hisat2官网下载index数据。当然也可以使用hisat2的组件hisat2-build来自己建立索引:
# 先通过注释文件建立含有剪接位点信息和exon信息的两个文件
# 其实hisat2-buld在运行的时候也会自己寻找exons和splice_sites,但是先做的目的是为了提高运行效率
extract_exons.py gencode.v26lift37.annotation.sorted.gtf > hg19.exons.gtf &
extract_splice_sites.py gencode.v26lift37.annotation.gtf > hg19.splice_sites.gtf &
# 建立index, 必须选项是基因组所在文件路径和输出的前缀
hisat2-build -p 4 --ss hg19.splice_sites.gtf --exon hg19.exons.gtf hg19.fa hg19
3.2 使用hisat2进行比对
建立好索引后便可以开始正式比对了:
#!/bin/bash
for i in `seq 56 62`;do
hisat2 -x /DataBase/Human/hg19/hg19_index/hg19/genome --threads 8 -1 ~/RNA/cleandata/SRR35899${i}_1.fastq.gz -2 ~/RNA/cleandata/SRR35899${i}_2.fastq.gz -S SRR35899${i}.sam
done
3.3 Reads Count
使用htseq进行reads计数。
#!/bin/bash
for i in `seq 56 58`;do
htseq-count -f sam -r pos -s no -a 10 -t exon -m union -i gene_id ~/RNA/mapping/SRR35899${i}.sam /DataBase/Human/hg19/encode_anno/gencode.v27lift37.annotation.gtf > SRR35899${i}.count
done
htseq-count运行完每个样本生成一个基因表达量文件,需要将所有样本合并起来进行表达差异分析,这里分别使用python以及linux命令进行简单的合并:
### Linux命令
$ paste *.count | awk 'BEGIN{FS="\t";OFS="\t";print "gene","c1","c2","c3"}{print $1,$2,$4,$6}' >merge.count
### Python, 这里用的pandas
import pandas as pd
df1 = pd.read_table('SRR3589956.count', header = None)
df2 = pd.read_table('SRR3589957.count', header = None)
df3 = pd.read_table('SRR3589958.count', header = None)
df1[2] = df2[1]
df1[3] = df3[1]
df1.columns = ['gene', 'SRR3589956', 'SRR3589957', 'SRR3589958']
df1.to_csv('merge.xlsx', sep='\t', index=False)