刘小泽写于19.12.29 + 2020.1.1
这一篇不扯别的,只列我认为比较重点的limma包的知识,所以代码部分可能比较多。但也会用不同的知识点来区分
前言
limma的全称是:Linear Models for Microarray Data
需要阅读limma的官方说明:https://www.bioconductor.org/packages/release/bioc/vignettes/limma/inst/doc/usersguide.pdf
尤其是第8章 Linear Models Overview
常用知识点
知识点一 (文字解释)
进行差异分析时常用limma。虽然它是针对芯片数据开发的,但也有limma-voom可以分析转录组数据,可以作为金标准。
它需要的输入文件有:
- 表达矩阵 (exprSet)(这个容易获得),芯片数据可以通过
exprs(),常规的转录组可以通过read.csv()等导入 - 分组矩阵 (design) :就是将表达矩阵的列(各个样本)分成几组(例如最简单的
case - control,或者一些时间序列的样本day0, day1, day2 ...)【通过model.matrix()得到】 - 比较矩阵(contrast):意思就是如何指定函数去进行组间比较【通过
makeContrasts()得到】
它的主要流程有:
- lmFit():线性拟合模型构建【需要两个东西:
exprSet和design】 ,得到的结果再和contrast一起导入contrasts.fit()函数 - eBayes():利用上一步
contrasts.fit()的结果 - topTable():利用上一步
eBayes()的结果,并最终导出差异分析结果
知识点二(代码演示)
搭配上面的解释来看,效果更好
分开展示 =》 构建三个输入文件
# 输入文件一:例如我现在已经有了一个表达矩阵eset
# 输入文件二:分组矩阵【假设9个样本分成了3组】
design <- model.matrix(~ 0+factor(c(1,1,1,2,2,3,3,3)))
colnames(design) <- c("group1", "group2", "group3")
rownames(design) <- colnames(eset)
# 输入文件三:比较矩阵【如果要进行三组之间的两两比较】
contrast.matrix <- makeContrasts(group2-group1, group3-group2, group3-group1, levels=design)
- 注意一个问题:样本数量和分组数量不一样,因为即使有100个样本,最后还可能依然分成2组(处理和对照组,我们只关心分组)
- 比较矩阵的组之间用
-连接
分开展示 =》 进行三个主要流程
# 第一步:lmFit
fit <- lmFit(eset, design)
fit2 <- contrasts.fit(fit, contrast.matrix)
# 第二步:eBayes
fit2 <- eBayes(fit2)
# 第三步:topTable【最后例如要挑出第一个比较组:group2-group1的差异分析结果】
topTable(fit2, coef=1, adjust="BH")
使用
coef参数,这里设为1,也就是表示根据上面makeContrasts的第一个(group2-group1)来提取结果-
adjust="BH"表示对p值的校正方法,包括了:"none","BH","BY"and"holm"。那么为啥要对P值进行校正呢?
p值是针对单次检验,假设采用的p值为小于0.05,我们通常认为这个基因在两组样本中的表达是有显著差异的,但是仍旧有5%的概率表示这个基因并不是差异基因。但是,当两组样本中有20000个基因采用同样的检验方式进行统计检验时,就会遇到一个问题:单次犯错的概率为0.05, 如果进行20000次检验,那么就会有0.05*20000=1000 个基因在组间的差异被错误估计
最后整合展示代码
# 还是假设有了表达矩阵eset
design <- model.matrix(~ 0+factor(c(1,1,1,2,2,3,3,3)))
colnames(design) <- c("group1", "group2", "group3")
fit <- lmFit(eset, design)
contrast.matrix <- makeContrasts(group2-group1, group3-group2, group3-group1, levels=design)
fit2 <- contrasts.fit(fit, contrast.matrix)
fit2 <- eBayes(fit2)
# 【例如要挑出第一个比较组:group2-group1的差异分析结果】
output <- topTable(fit2, coef=1, adjust="BH")
最后的最后,别忘了去掉那些NA值
limma_DEG = na.omit(output)
# 然后可以选择保存
# write.csv(limma_DEG,"limma_results.csv",quote = F)
知识点三 一定要使用比较矩阵吗?
答案是不一定
看这里:https://github.com/bioconductor-china/basic/blob/master/makeContrasts.md
然后可以再结合说明书Chapter9.2 (第42页)
Group <- factor(targets$Target, levels=c("WT","Mu"))
-
如果使用:
design <- model.matrix(~Group) colnames(design) <- c("WT","MUvsWT")那么它已经把要比较的组放在了第一列,然后其余的列都与第一列进行比较,而结果使用
coef进行指定提取 -
或者可以用
design <- model.matrix(~0+Group) colnames(design) <- c("WT","MU")它没有设置默认如何比较,仅仅是做了一个分组,后续还需要使用
makeContrasts来定义
其实差异分析,一个使用难点就是:分组
limma包关于如何针对特殊情况进行分组描述了很大的篇幅
例如 如果包含多个组多个处理
参考:limma说明书 P43
-
实验设计背景
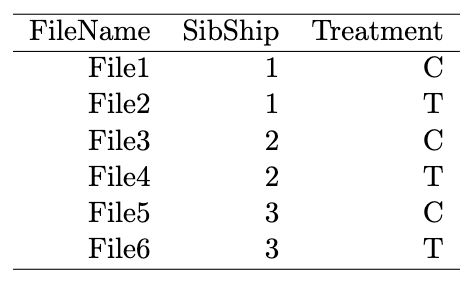 如果包含多个组多个处理
如果包含多个组多个处理有6个样本,分成3组(1、2、3),并且每组包括两个处理方式:一个对照(C),一个处理(T)
-
我们自己来构建这样一个数据
targets <- data.frame(FileName=paste0("File",1:6), SibShip=c(1,1,2,2,3,3), Treatment=rep(c("C","T"),3)) -
分组矩阵这样设计
library(limma) > (SibShip <- factor(targets$SibShip)) [1] 1 1 2 2 3 3 Levels: 1 2 3 > (Treat <- factor(targets$Treatment, levels=c("C","T"))) [1] C T C T C T Levels: C T # 注意这种设计方式: design <- model.matrix(~SibShip+Treat) -
最后还是三步走
fit <- lmFit(eset, design) fit <- eBayes(fit) topTable(fit, coef="TreatT")
上面分组矩阵的设计规律就是:
design <- model.matrix(~Block+Treatment)
将每个组作为一个block,其中只比较组内的处理和对照(Treatment)
例如 时间顺序的样本
参考P47 的 Chapter 9.6
比方说,有两种基因型(wt、mu),各自测了3个时间点(0、6、24h)

可以这样操作:
lev <- c("wt.0hr","wt.6hr","wt.24hr","mu.0hr","mu.6hr","mu.24hr")
f <- factor(Target, levels=lev)
design <- model.matrix(~0+f)
colnames(design) <- lev
fit <- lmFit(eset, design)
如果要探索野生型中从0到6、从6到24h等待后发生了什么变化
cont.wt <- makeContrasts(
"wt.6hr-wt.0hr",
"wt.24hr-wt.6hr", levels=design)
fit2 <- contrasts.fit(fit, cont.wt)
fit2 <- eBayes(fit2)
topTableF(fit2, adjust="BH")
# 那么对于mu型也是一样的
如果要探索从0-6、从6到24h处理后,mu相对wt发生了什么变化
cont.dif <- makeContrasts(
Dif6hr =(mu.6hr-mu.0hr)-(wt.6hr-wt.0hr),
Dif24hr=(mu.24hr-mu.6hr)-(wt.24hr-wt.6hr),
levels=design)
fit3 <- contrasts.fit(fit, cont.dif)
fit3 <- eBayes(fit3)
topTableF(fit3, adjust="BH")
当然,还有很多很多的例子,都在说明书有体现。
这里只是列出来一个思路:凡是想用一个包,它的帮助文档就是最好的答案
欢迎关注我们的公众号~_~
我们是两个农转生信的小硕,打造生信星球,想让它成为一个不拽术语、通俗易懂的生信知识平台。需要帮助或提出意见请后台留言或发送邮件到[email protected]
