Introduction 介绍
GDC mRNA定量分析管道测量 HT-Seq 原始reads统计中的基因表达水平,Fragments per Kilobase of transcript per Million mapped reads(FPKM)和FPKM-UQ(上四分位标准化)。 首先将reads与GRCh38 reference genome 参考基因组比对,然后通过量化映射的reads产生这些值。 为了促进样品间归一化,所有RNA-Seq读数在分析过程中都被视为unstranded的状态.
Data Processing Steps 数据处理步骤
RNA-Seq 比对流程
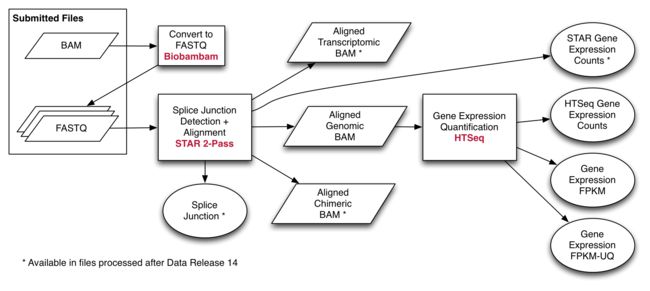
以 Alignment Workflow 开始比对的流程, 该流程使用 STAR 中重复比对方法执行. STAR 分别比对每个 read group 然后将得到的比对文件合并为一个。按照国际癌症基因组协会 ICGC ( github) 使用的方法, the two-pass method 包含剪接点检测步骤,其用于产生最终比对。 此工作流程输出基因组BAM文件,其中包含比对和未比对的reads。 质量评估在比对前用 FASTQC 进行,并在比对后用 Picard Tools 进行。.
除了上面详述的基因组比对之外,在数据发布之后处理的文件具有相关的转录组和嵌合比对。这仅用于至少有1组paired-end reads的等份样品. 嵌合的BAM文件包含mapping到不同染色体或链的reads(融合比对)。 基因组比对文件包含嵌合和未对齐的reads,以便于检索所有原始reads。 转录组比对报告使用转录物坐标而不是基因组坐标比对reads。转录组比对队列也被不同地排序以促进下游分析。 这种排序方法不允许在这些排列上进行BAM切片,故不支持BAM索引文件配对。 这些对齐的拼接头文件也可用。
| I/O | Entity | Format |
|---|---|---|
| Input | Submitted Unaligned Reads or Submitted Aligned Reads | FASTQ or BAM |
| Output | Aligned Reads | BAM |
RNA-Seq Alignment 命令行参数
请注意,由于正在进行管道开发和改进,从GDC门户下载的文件中的版本号可能会有所不同。
- Original
- Dr15plus
# STAR-2.4.2a
### For users with access to the ICGC pipeline:
python star_align.py \
--genomeDir \
--FastqFileIn \
--workDir \
--out \
--genomeFastaFiles \
--runThreadN 8 \
--outFilterMultimapScoreRange 1 \
--outFilterMultimapNmax 20 \
--outFilterMismatchNmax 10 \
--alignIntronMax 500000 \
--alignMatesGapMax 1000000 \
--sjdbScore 2 \
--limitBAMsortRAM 0 \
--alignSJDBoverhangMin 1 \
--genomeLoad NoSharedMemory \
--outFilterMatchNminOverLread 0.33 \
--outFilterScoreMinOverLread 0.33 \
--twopass1readsN -1 \
--sjdbOverhang 100 \
--outSAMstrandField intronMotif \
--outSAMunmapped Within
### For users without access to the ICGC pipeline:
### Step 1: Building the STAR index.*
STAR
--runMode genomeGenerate
--genomeDir
--genomeFastaFiles
--sjdbOverhang 100
--sjdbGTFfile
--runThreadN 8
### Step 2: Alignment 1st Pass.
STAR
--genomeDir
--readFilesIn ,,... ,,...
--runThreadN
--outFilterMultimapScoreRange 1
--outFilterMultimapNmax 20
--outFilterMismatchNmax 10
--alignIntronMax 500000
--alignMatesGapMax 1000000
--sjdbScore 2
--alignSJDBoverhangMin 1
--genomeLoad NoSharedMemory
--readFilesCommand
--outFilterMatchNminOverLread 0.33
--outFilterScoreMinOverLread 0.33
--sjdbOverhang 100
--outSAMstrandField intronMotif
--outSAMtype None
--outSAMmode None
### Step 3: Intermediate Index Generation.
STAR
--runMode genomeGenerate
--genomeDir
--genomeFastaFiles
--sjdbOverhang 100
--runThreadN
--sjdbFileChrStartEnd
### Step 4: Alignment 2nd Pass.
STAR
--genomeDir
--readFilesIn ,,... ,,...
--runThreadN
--outFilterMultimapScoreRange 1
--outFilterMultimapNmax 20
--outFilterMismatchNmax 10
--alignIntronMax 500000
--alignMatesGapMax 1000000
--sjdbScore 2
--alignSJDBoverhangMin 1
--genomeLoad NoSharedMemory
--limitBAMsortRAM 0
--readFilesCommand
--outFilterMatchNminOverLread 0.33
--outFilterScoreMinOverLread 0.33
--sjdbOverhang 100
--outSAMstrandField intronMotif
--outSAMattributes NH HI NM MD AS XS
--outSAMunmapped Within
--outSAMtype BAM SortedByCoordinate
--outSAMheaderHD @HD VN:1.4
--outSAMattrRGline
*这些索引可在 GDC Website 上下载,无需再次构建。
mRNA 表达量处理流程
比对后,通过 RNA Expression Workflow 处理BAM文件以确定RNA表达水平。
映射到每个基因的读数使用HT-Seq-Count计数。表达式值以制表符分隔的格式提供。 GENCODE v22 用于基因注释。
在Data Release 14之后处理的文件具有STAR在对齐步骤期间生成的额外读取计数集。
| I/O | Entity | Format |
|---|---|---|
| Input | Aligned Reads | BAM |
| Output | Gene Expression | TXT |
mRNA Quantification 命令行参数
HTSeq-0.6.1p1
- Original
- Dr15plus
htseq-count \
-m intersection-nonempty \
-i gene_id \
-r pos \
-s no \
- gencode.v22.annotation.gtf
mRNA Expression HT-Seq Normalization 表达标准化
由HT-Seq产生的RNA-Seq表达水平reads计数使用两种类似的方法标准化:FPKM和FPKM-UQ。标准化值应仅在整个基因集的上下文中使用。如果研究了一组基因,鼓励用户将原始reads计数值标准化。
FPKM
The Fragments per Kilobase of transcript per Million mapped reads (FPKM) 计算通过将读数除以基因长度和映射到蛋白质编码基因的读数总数来标准化读数。
Upper Quartile FPKM
The upper quartile FPKM (FPKM-UQ) 是一种修改的FPKM计算,其中总蛋白质编码读数计数被样品的第75百分位读数计数值代替。
Calculations
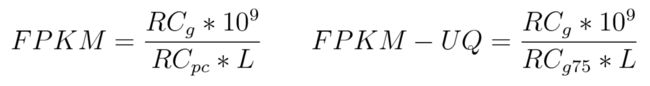
- RCg: 映射到Gene的reads数
- RCpc: 映射到所有蛋白质编码基因的reads数
- RCg75: 本中基因的第75百分位reads计数值
- L: Length of the gene in base pairs; 计算为基因中所有外显子的总和
Note: 在归一化时,reads计数乘以标量(109) 以考虑千碱基和'百万映射读数'单位
Examples 样品
Sample 1: Gene A
- Gene length: 3,000 bp
- 1,000 reads mapped to Gene A
- 1,000,000 reads mapped to all protein-coding regions
- Read count in Sample 1 for 75th percentile gene: 2,000
FPKM for Gene A = (1,000)(10^9)/[(3,000)(1,000,000)] = 333.33
FPKM-UQ for Gene A = (1,000)(10^9)/[(3,000)(2,000)] = 166,666.67
File Access and Availability 文件访问和可用性
为了便于在用户创建的管道中使用协调数据,可以在GDC数据门户中的几个中间步骤中访问RNA-Seq基因表达。以下是可在GDC Data Portal中下载的每种文件类型的说明。
| Type | Description | Format |
|---|---|---|
| RNA-Seq Alignment | 已经与GRCh38构建一致的RNA-Seq reads。包括未比对上的reads以促进原始读取集的可用性 | BAM |
| HT-Seq Read Counts | 通过HT-Seq计算的与每个基因比对的reads数目 | TXT |
| STAR Read Counts | STAR计算的比对到每个基因的reads数 | TSV |
| FPKM | 标准化的表达值,其考虑每个基因长度和映射到所有蛋白质编码基因的reads的数量 | TXT |
| FPKM-UQ | FPKM公式的修改版本,其中第75百分位reads计数用作分母代替蛋白质编码的总reads数 | TXT |