之前写过ChIP-seq的基础分析:ChIP-seq数据分析(一):从raw reads到peaks,和此文有很多相似之处。
简介
ATAC-seq的目的是鉴定DNA的可触及区域,即未被组蛋白保护的开放区域,相当于DNase I 能够起作用的区域。用来探究转录因子反式调控机制。
参考:
ATAC-seq测序的原理及应用http://www.360doc.com/content/18/1221/00/52645714_803259507.shtml
0. 参考基因组建立索引
bowtie2-build -f Arabidopsis_thaliana.TAIR10.dna.toplevel.fa TAIR10.bybowtie2
1. 下载数据并解压
for sample in SRR58746{57..62}
do
prefetch $sample
done
ls *sra | while read id; do fastq-dump --gzip --split-3 --defline-qual '+' --defline-seq '@$ac-$si/$ri' ${id} & done
2. 数据质控
cd ~/ATAC_seq/raw
mkdir fastqc_report
fastqc *fastq.gz -o ./fastqc_report/
cd fastqc_report/
~/miniconda3/bin/multiqc ./
安装multiqc
git clone https://github.com/ewels/MultiQC.git
~/miniconda3/bin/python3 setup.py install
~/miniconda3/bin/multiqc
从文献中可以看到测的是paired-end 36 nt reads。在经过fastqc检测之后发现,不含接头且测序质量很高,有一个样本的一端reads中含有1/1000的overrepresented sequences,没有做处理。
3. 比对
cd ~/ATAC_seq/align
for i in SRR58746{57..62}
do
bowtie2 -x /ifs1/Grp3/huangsiyuan/Ref/Arabidopsis/TAIR10.bybowtie2 -X 1000 -1 /ifs1/Grp3/huangsiyuan/ATAC_seq/raw/${i}_1.fastq.gz -2 /ifs1/Grp3/huangsiyuan/ATAC_seq/raw/${i}_2.fastq.gz | samtools view -F 4 -bS - > ./${i}.bam &
done
在上述比对命令中,
-F 4表示:只要flag中(是一个累加值)含有4,就不要这一个reads比对记录;4表示:read unmapped (0x4)
下文中有-f的用法,
-f 2表示:只要flag中(是一个累加值)含有2,就保留这个比对记录;2表示:read mapped in proper pair (0x2)
关于sam文件的flag值可以在这里查询到,
http://broadinstitute.github.io/picard/explain-flags.html
4. 比对之后的质控
cd ~/ATAC_seq/align
for i in SRR58746{57..62}
do
samtools view -f 2 -q 30 -o ${i}.f2.q30.bam ${i}.bam #注1
samtools sort -@ 4 -o ${i}.f2.q30.sort.bam ${i}.f2.q30.bam
java -jar /ifs1/Software/biosoft/picard/picard.jar MarkDuplicates VERBOSITY=ERROR QUIET=true \
CREATE_INDEX=false REMOVE_DUPLICATES=true \
INPUT=${i}.f2.q30.sort.bam \
OUTPUT=${i}.f2.q30.sort.rmdup.bam \
M=${i}.marked_dup.log
samtools view -h ${i}.f2.q30.sort.rmdup.bam | grep "Mt" -v | grep "Pt" -v | samtools view -bS -o ${i}.final.bam
done
注1
此时bam文件的第二列flag值只有4种
99: 0x1; 0x2; 0x20; 0x40(都是16进制)
147: 0x1; 0x2; 0x10; 0x80
83: 0x1; 0x2; 0x10; 0x40
163: 0x1; 0x2; 0x20; 0x80
我的理解
0x1:read paired,表示双端测序,并且两条reads都在比对记录中
0x2:read mapped in proper pair,表示这条read比对上了,它的另一条read也比对上,并且它俩距离合适
0x10:read reverse strand,这条read比对到负链
0x20:mate reverse strand,这条read对应的另一条read比对到负链(一般都是一条比对到正链,另一条比对到负链)
0x40:first in pair,一对reads中的第一条
0x80:second in pair,一对reads中的第二条(根据read的名称来判断,如fastq文件中reads名称的/1、/2,这个标识会在比对之后省略)
5. 插入片段长度分布
- 什么叫insert size?参考《一篇文章说清楚什么是“插入片段”》——公众号“碱基矿工”
- 如何从sam文件中求得insert size:在“read mapped in proper pair”的前提下,取第九列
cd ~/ATAC_seq/align
for i in SRR58746{57..62}
do
samtools view -f 64 ${i}.final.bam | perl -ane 'print abs($F[8]);print "\n";' | sort -n > ${i}.insert_size.txt
done
这里-f 64的64就是0x40,表示提取一半的比对记录,因为两条reads对应一个insert size。
在RStudio上运行
df <- read.table("SRR5874659.insert_size.txt",header = F)
hist(df$V1,main = "insert size distribution",ylab = "read count",xlab = "insert size",breaks = seq(0,max(df$V1),by = 1), col = "lightblue",border="lightblue")
axis(side = 1,at=seq(0,max(df$V1),by = 100),labels = seq(0,max(df$V1),by = 100))

6. Peak Calling
Chip-seq与ATAC-seq call peaks的区别,前者peaks是代表抗体结合转录因子的位点,后者peaks是代表Tn5转座酶切开染色质开放区的两端,因此在一个位置,前者peaks有一个,后者有两个
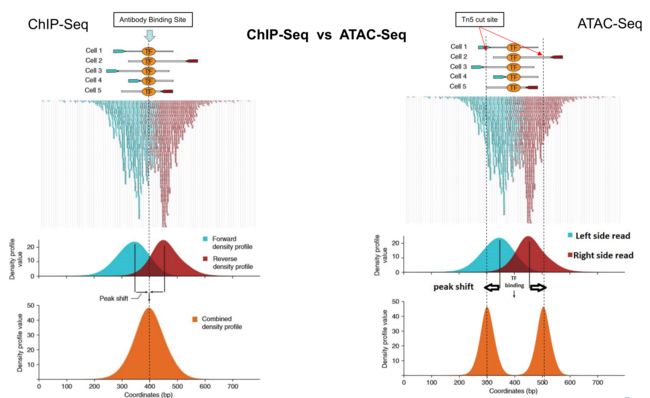
6.1 bamtobed
for i in SRR58746{57..62}
do
bedtools bamtobed -i ${i}.final.bam > ${i}.final.bed
done
这里需要留意一下bam和bed两种文件的起始坐标,分别是以1、0开始的
1 0 35 SRR5874657-27764514/2 42 +
1 13 48 SRR5874657-32600261/2 42 +
1 18 54 SRR5874657-18292360/1 42 +
6.2 call peaks
cd ~/ATAC_seq/peak/
for i in SRR58746{57..62}
do
macs2 callpeak -t ~/ATAC_seq/align/${i}.final.bed -n ${i} --shift -75 --extsize 150 --nomodel -B --SPMR -g 110000000 --keep-dup all
done
关于结果的解读,参考:https://www.jianshu.com/p/e83a7e10ea2e
7. bedGraphToBigWig
之前ChIP-seq里面讲过deeptools来转格式bam2bw,现在学的是bdg2bw,是一回事吗?
这一步使用到的工具:bedSort bedClip bedGraphToBigWig。下载:http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/bedSort
http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/bedClip
http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/bedGraphToBigWig
下载之后,赋予权限,再转移到一个PATH包含的目录下,
mv bedSort bedClip bedGraphToBigWig ~/local/bin/
#求染色体序列大小
samtools view -H SRR5874657.final.bam | perl -ne 'if(/SN:(\S+)\s+LN:(\d+)/){print "$1\t$2\n"}' > chr.size
for i in SRR58746{57..62}
do
#对treat_pileup.bdg文件的前三列排序,原文件的染色体顺序不定
~/local/bin/bedSort ${i}_treat_pileup.bdg ${i}_treat_pileup.sort.bdg
#根据染色体的最大坐标,修正部分记录的第三列(原文件的这些记录的第三列大于max_chr_pos,原因是前面在call peaks的时候reads有偏移);
#这样一来,这些记录的第二列就比第三列大了,需要过滤掉
~/local/bin/bedClip -truncate ${i}_treat_pileup.sort.bdg ~/ATAC_seq/align/chr.size stdout | perl -ane 'print if($F[1] < $F[2])' > ${i}_treat_pileup.bedgrafh
#以上两步的目的是生成一个“可读性”良好的bedgrafh文件,与原bedgrafh文件有区别
#bedgraph to bigwig
~/local/bin/bedGraphToBigWig ${i}_treat_pileup.bedgrafh ~/ATAC_seq/align/chr.size ${i}_treat_pileup.bw
done
8. 可视化
8.1 TSS Enrichment
TSS富集结果依赖于基因注释结果。这个图在学习ChIP-seq的时候也画过,方法一样
cd ~/ATAC_seq/visual
for i in SRR58746{57..62}
do
~/miniconda3/bin/computeMatrix reference-point -S ~/ATAC_seq/peak/${i}_treat_pileup.bw -R ~/Ref/Arabidopsis/Arabidopsis_thaliana.TAIR10.44.gtf -a 3000 -b 3000 -p 1 -o ${i}.matrix.TSS.3k.gz
~/miniconda3/bin/plotHeatmap -m ${i}.matrix.TSS.3k.gz -o ${i}.TSS.3k.png --colorMap Reds
done
8.2 IGV中查看数据
导入bigwig文件和narrowPeak文件到IGV中(可以显示peaks的名称),即可查看ATAC-Seq数据的结果。这一步之前也做过
9. 多样品数据可重复评估
9.1 用bedtools计算overlap
wc -l SRR587465{7..9}_peaks.narrowPeak
30123 SRR5874657_peaks.narrowPeak
30644 SRR5874658_peaks.narrowPeak
30806 SRR5874659_peaks.narrowPeak
bedtools intersect -a SRR5874657_peaks.narrowPeak -b SRR5874658_peaks.narrowPeak -wa > tmp
bedtools intersect -a tmp -b SRR5874659_peaks.narrowPeak -wa | wc -l && rm -f tmp
29310
wc -l SRR587466{0..2}_peaks.narrowPeak
23965 SRR5874660_peaks.narrowPeak
26038 SRR5874661_peaks.narrowPeak
24812 SRR5874662_peaks.narrowPeak
bedtools intersect -a SRR5874660_peaks.narrowPeak -b SRR5874661_peaks.narrowPeak -wa > tmp
bedtools intersect -a tmp -b SRR5874662_peaks.narrowPeak -wa | wc -l && rm -f tmp
22773
由以上可知,重复性不错。
9.2 IDR评估
同时考虑peaks间的overlap和富集倍数的一致性。
使用和结果解读参考:https://www.jianshu.com/p/d8a7056b4294
#安装
~/miniconda3/bin/conda install idr
#先根据-log10(p-value)进行排序
for i in SRR58746{57..62}
do
sort -k8,8nr ~/ATAC_seq/peak/${i}_peaks.narrowPeak > ~/ATAC_seq/idr/${i}_peaks_sort.narrowPeak
done
#使用示例
~/miniconda3/bin/idr --samples SRR5874657_peaks_sort.narrowPeak SRR5874658_peaks_sort.narrowPeak -o SRR5874657_SRR5874658_idr --plot --input-file-type narrowP
eak --rank p.value &
会生成两个文件:
SRR5874657_SRR5874658_idr.png
SRR5874657_SRR5874658_idr

没有通过特定IDR阈值的peaks显示为红色
wc -l SRR5874657_SRR5874658_idr
28433 #共有的部分
awk '{if($5 >= 540) print $0}' SRR5874657_SRR5874658_idr | wc -l
19317 #满足IDR的部分
19317/28433=67.9%
10. 注释
这一步前面ChIP-seq也做了,感觉差不多
library(ChIPseeker)
library(GenomicFeatures)
ath <- makeTxDbFromGFF("E:/Computational_Biologist/生信积累/测序类/表观遗传/ChIP-seq/拟南芥ChIP-seq_BMC基因组/ref/Arabidopsis_thaliana.TAIR10.44.gff3")
peaksfile <- "SRR5874658_peaks.narrowPeak"
peak <- readPeakFile(peaksfile,header=F)
outname="SRR5874658"
peakAnno <- annotatePeak(peak,TxDb = ath,assignGenomicAnnotation = TRUE)
pdf(file = paste0(outname,"peakAnnotation.pdf"))
plotAnnoPie(peakAnno,main=paste0(outname,"\nDistribution of Peaks"),line=-8)
dev.off()
#保存peaks的注释文件方便改图
write.table(as.data.frame(peakAnno@anno),file = paste0(outname,"peakAnnotation.txt"),sep = "\t",row.names = FALSE)
