生信人的20个R语言习题
1 安装R包
数据包: ALL, CLL, pasilla, airway
软件包:limma,DESeq2,clusterProfiler
工具包:reshape2
绘图包:ggplot2
options()$repos
options()$BioC_mirror
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")
options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(c("ALL","CLL","pasilla", "airway") ,ask = F,update = F)
BiocManager::install(c("limma","DESeq2", "clusterProfiler") ,ask = F,update = F)
install.packages("reshape2") #工具包
install.packages("ggplot2") #绘图包
2 了解ExpressionSet对象
CLL包里面就有data(sCLLex) ,找到它包含的元素,提取其表达矩阵(使用exprs函数),查看其大小
- 参考:http://www.bio-info-trainee.com/bioconductor_China/software/limma.html
- 参考:https://github.com/bioconductor-china/basic/blob/master/ExpressionSet.md
suppressPackageStartupMessages(library(CLL)) #隐藏具体加载信息
data(sCLLex)
exprSet=exprs(sCLLex) ##sCLLex是依赖于CLL这个package的一个对象
samples=sampleNames(sCLLex)
pdata=pData(sCLLex)
group_list=as.character(pdata[,2])
dim(exprSet)
3 了解 str,head,help函数,作用于第二步提取到的表达矩阵
str(exprSet)
head(exprSet)
help("exprSet")
4 安装并了解hgu95av2.db包
安装并了解 hgu95av2.db 包,看看 ls("package:hgu95av2.db") 后显示的那些变量
BiocManager::install("hgu95av2.db")
library(hgu95av2.db)
ls("package:hgu95av2.db")

5.理解 head(toTable(hgu95av2SYMBOL)) 的用法
理解 head(toTable(hgu95av2SYMBOL)) 的用法,找到 TP53 基因对应的探针ID
head(toTable(hgu95av2SYMBOL))
probe_id symbol
1 1000_at MAPK3
2 1001_at TIE1
3 1002_f_at CYP2C19
4 1003_s_at CXCR5
5 1004_at CXCR5
6 1005_at DUSP1
ids=toTable(hgu95av2SYMBOL)
#在ids中搜索TP53
> ids[which(ids[,2]=="TP53"),]#记住
probe_id symbol
966 1939_at TP53
997 1974_s_at TP53
1420 31618_at TP53
> ids[grep("^TP53$",ids$symbol),]#记住
probe_id symbol
966 1939_at TP53
997 1974_s_at TP53
1420 31618_at TP53

6.理解探针与基因的对应关系
理解探针与基因的对应关系,总共多少个基因,基因最多对应多少个探针,是哪些基因,是不是因为这些基因很长,所以在其上面设计多个探针呢?
summary(hgu95av2SYMBOL)
SYMBOL map for chip hgu95av2 (object of class "ProbeAnnDbBimap")
|
| Lkeyname: probe_id (Ltablename: probes)
| Lkeys: "1000_at", "1001_at", ... (total=12625/mapped=11460)
|
| Rkeyname: symbol (Rtablename: gene_info)
| Rkeys: "A1BG", "A2M", ... (total=61050/mapped=8585)
|
| direction: L --> R
#左边: 探针总数是12625,能匹配上的是11460个;
#右边: 基因名的总数是30071,实际上只有8585种
#或者
mapped_probes <- mappedkeys(hgu95av2SYMBOL) #手动过滤,然后进行统计
count.mappedkeys(hgu95av2SYMBOL) #统计:与summary结果相符
[1] 11460
#有1165个探针没有匹配上基因。(12625-11460=1165)
ids <- toTable(hgu95av2SYMBOL)
1.dim(ids)
[1] 11460 2
2.length(unique(ids$symbol))
[1] 8585
3.tail(sort(table(ids$symbol)))
YME1L1 GAPDH INPP4A MYB PTGER3 STAT1
7 8 8 8 8 8
4.table(sort(table(ids$symbol)))#sort(table(ids$symbol))统计的是每个基因对应的探针数目。
1 2 3 4 5 6 7 8
6555 1428 451 102 22 16 6 5
##一个探针对应唯一一个基因的为6555个探针,也就是6555个基因是对应唯一一个探针的。还有一个基因对应多个探针的,比如最后一列,就是一个基因对应8个探针,这样的基因共有5个。
#所以其实6555+1428+451+102+22+16+6+5=8585,1428、451等是基因数目,只不过对应的探针数目不同而已,但也是unique的,因此同样应该是算在unique(ids$symbol)里面的。
7.找到那些不在 hgu95av2.db 包收录的对应着SYMBOL的探针。
第二步提取到的表达矩阵是12625个探针在22个样本的表达量矩阵,找到那些不在 hgu95av2.db 包收录的对应着SYMBOL的探针。
从上一步了解到有1165个探针是不能匹配上基因名字的,现在就是要找出这些探针。
#第二步获得的exprSet这个表达矩阵
dim(exprSet)
[1] 12625 22
#以下为e1和e2在 hgu95av2.db 包收录的对应着SYMBOL的探针。
e1<-exprSet[rownames(exprSet) %in% ids$probe_id,]
dim(e1)
[1] 11460 22
e2<-exprSet[match(rownames(exprSet),ids$probe_id, nomatch = 0),]
dim(e2)
[1] 11460 22
#但要找1165个不能匹配上基因名字的探针
e3<-exprSet[!rownames(exprSet) %in% ids$probe_id,]
dim(e3)
[1] 1165 22
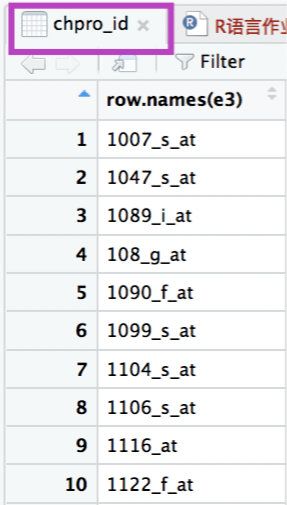
chpro_id<-as.data.frame(row.names(e3))
> dim(chpro_id)
[1] 1165 1
8.过滤表达矩阵,删除那1165个没有对应基因名字的探针。
#思路是不删除,直接取有基因名字的探针为一个表达矩阵。同第7题中的e1和e2。
e1<-exprSet[rownames(exprSet) %in% ids$probe_id,]
e2<-exprSet[match(rownames(exprSet),ids$probe_id, nomatch = 0),]
9.整合表达矩阵
整合表达矩阵,多个探针对应一个基因的情况下,只保留在所有样本里面平均表达量最大的那个探针。
提示,理解 tapply,by,aggregate,split 函数 , 首先对每个基因找到最大表达量的探针,然后根据得到探针去过滤原始表达矩阵。
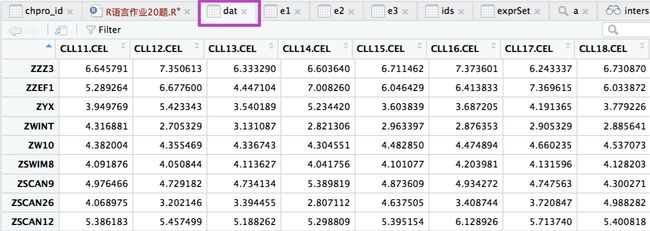
#新建dat
dat=exprSet[rownames(exprSet) %in% ids$probe_id,]
ids$median=apply(dat,1,median)#对dat每一行求median值
ids=ids[order(ids$symbol,ids$median,decreasing = T),]#order是按从小到达排序,加了decreasing = T是按从大到小排序。ids$symbol是将symbol基因名按照字母顺序从最后即"z"排序的,然后每个z开头的再按ids$median排序
ids=ids[!duplicated(ids$symbol),]#将ids$symbol去重后作为新的行,ids取这些行
dat=dat[ids$probe_id,]
dim(dat)
[1] 8585 22 #此时这个8585就是前面第6题中unique(ids$symbol)的基因的数目
10.把过滤后的表达矩阵更改行名为基因的symbol,因为这个时候探针和基因是一对一关系了。
rownames(dat)<-ids$symbol
11.对第10步得到的表达矩阵进行探索
对第10步得到的表达矩阵进行探索,先画第一个样本的所有基因的表达量的boxplot,hist,density,然后画所有样本的这些图。
参考:http://bio-info-trainee.com/tmp/basic_visualization_for_expression_matrix.html 理解ggplot2的绘图语法,数据和图形元素的映射关系。
##需要加载以下包
library(CLL)
library(ggplot2)
library(reshape2)
library(gpairs)
library(corrplot)
##把我们的宽矩阵用reshape2包变成长矩阵,用melt这个函数
library(reshape2)
exprSet<-dat #后面的代码是exprSet,因此又转换成exprSet
exprSet_L=melt(exprSet)#后面的矩阵都为exprSet_L
colnames(exprSet_L)=c('probe','sample','value')
exprSet_L$group=rep(group_list,each=nrow(dat))
head(exprSet_L)
probe sample value group
1 ZZZ3 CLL11.CEL 6.645791 progres.
2 ZZEF1 CLL11.CEL 5.289264 progres.
3 ZYX CLL11.CEL 3.949769 progres.
4 ZWINT CLL11.CEL 4.316881 progres.
5 ZW10 CLL11.CEL 4.382004 progres.
6 ZSWIM8 CLL11.CEL 4.091876 progres.
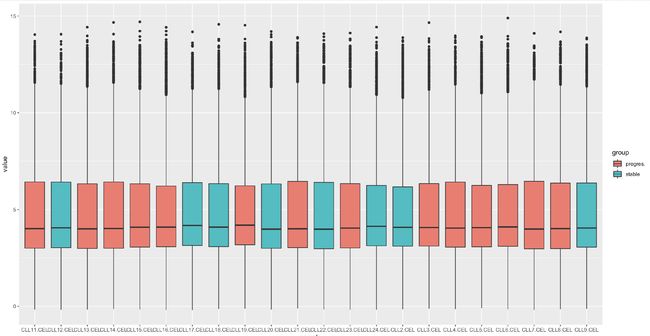
p=ggplot(exprSet_L,aes(x=sample,y=value,fill=group))+geom_boxplot()
print(p)
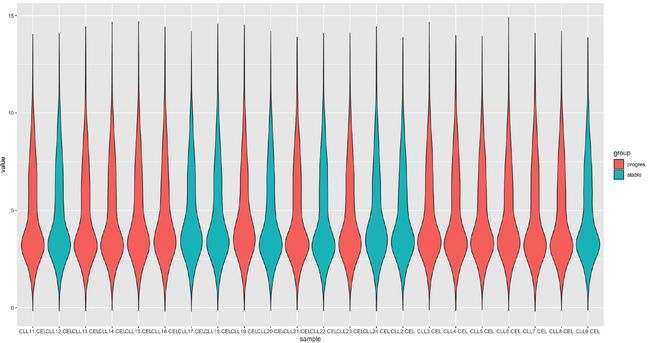
p=ggplot(exprSet_L,aes(x=sample,y=value,fill=group))+geom_violin()
print(p)
p=ggplot(exprSet_L,aes(value,fill=group))+geom_histogram(bins = 200)+facet_wrap(~sample, nrow = 4)
print(p)
p=ggplot(exprSet_L,aes(value,col=group))+geom_density()+facet_wrap(~sample, nrow = 4)
print(p)

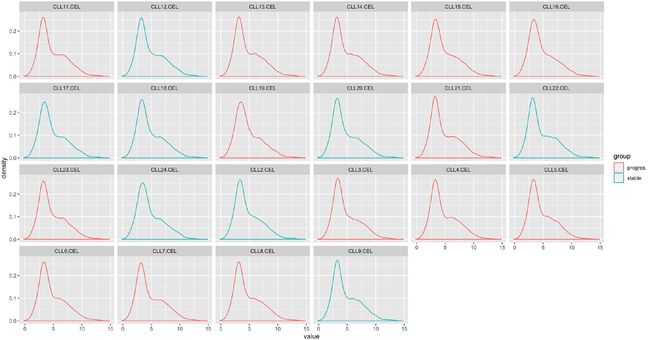
12.理解统计学指标mean,median,max,min,sd,var,mad并计算出每个基因在所有样本的这些统计学指标,最后按照mad值排序,取top 50 mad值的基因,得到列表。
e_mean = tail(sort(apply(exprSet,1,mean)),30)
e_median = tail(sort(apply(exprSet,1,median)), 30)
e_max <- tail(sort(apply(exprSet,1,max)),30)
e_min <- tail(sort(apply(exprSet,1,min)),30)
e_sd <- tail(sort(apply(exprSet,1,sd)),30)
e_var <- tail(sort(apply(exprSet,1,var)),30)
e_mad <- tail(sort(apply(exprSet,1,mad)),30) #绝对中位差来估计方差,先计算出数据与它们的中位数之间的偏差,然后这些偏差的绝对值的中位数就是mad
13.根据第12步骤得到top 50 mad值的基因列表来取表达矩阵的子集,并且热图可视化子表达矩阵。试试看其它5种热图的包的不同效果。
dat_top50<-dat[tail(sort(apply(exprSet,1,mad)),50),] #图左
choose_gene=names(sort(apply(exprSet, 1, mad),decreasing = T)[1:50])
choose_matrix=exprSet[choose_gene,]
choose_matrix=scale(choose_matrix)
heatmap(choose_matrix)
library(gplots) #图右上
heatmap.2(choose_matrix)
library(pheatmap) #图右下
choose_gene=names(tail(sort(apply(exprSet,1,mad)),50))
choose_matrix=exprSet[choose_gene,]
choose_matrix=t(scale(t(choose_matrix)))
pheatmap(choose_matrix)

14.取不同统计学指标mean,median,max,mean,sd,var,mad的各top50基因列表,使用UpSetR包来看他们之间的overlap情况。
library(UpSetR)
g_all <- unique(c(names(e_mean),names(e_median),names(e_max),names(e_min),
names(e_sd),names(e_var),names(e_mad) ))
dat=data.frame(e_all=g_all,
e_mean=ifelse(g_all %in% names(e_mean) ,1,0),
e_median=ifelse(g_all %in% names(e_median) ,1,0),
e_max=ifelse(g_all %in% names(e_max) ,1,0),
e_min=ifelse(g_all %in% names(e_min) ,1,0),
e_sd=ifelse(g_all %in% names(e_sd) ,1,0),
e_var=ifelse(g_all %in% names(e_var) ,1,0),
e_mad=ifelse(g_all %in% names(e_mad) ,1,0)
)
upset(dat,nsets = 7)
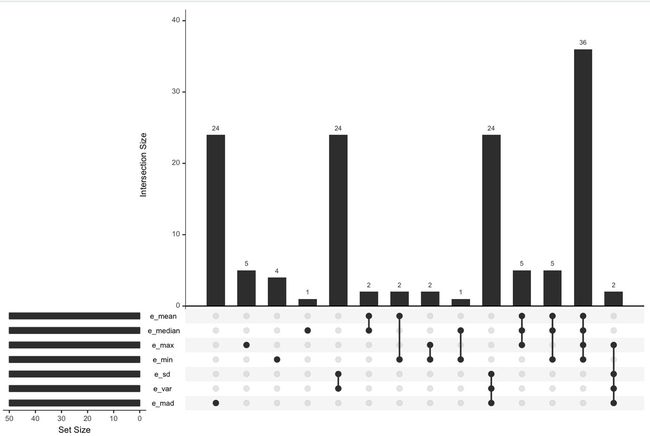
15.在第二步的基础上面提取CLL包里面的data(sCLLex) 数据对象的样本的表型数据。
pd = pData(sCLLex)
group_list = as.character(pd[,2])
table(group_list)
group_list
progres. stable
14 8
16.对所有样本的表达矩阵进行聚类并且绘图,然后添加样本的临床表型数据信息(更改样本名)
out.dist=dist(t(exprSet),method='euclidean') #用dist这个函数计算样本间的距离,这里的t(exprSet)是因为样本要在行,而目前的exprSet是样本在列,基因名字在行,所以t(exprSet)将行和列换位置
out.hclust=hclust(out.dist,method='complete')
plot(out.hclust)
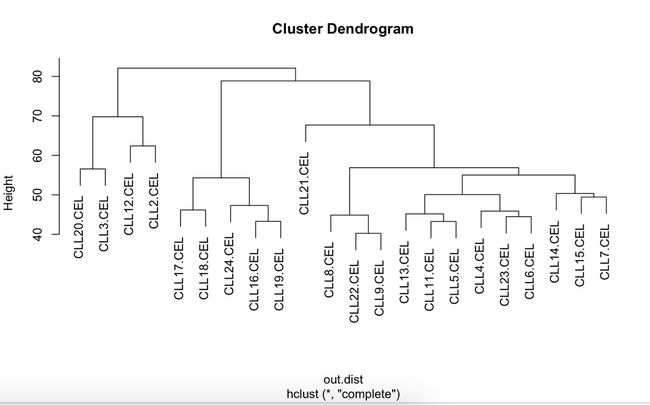
out.dist=dist(t(exprSet),method='euclidean')
out.hclust=hclust(out.dist,method='ward.D2')
plot(out.hclust)

可以在
hclust()里面调整参数method,选择不同的方法:hclust(E.dist, method="single") # 最近法
hclust(E.dist, method="complete") # 最远法
hclust(E.dist, method="average") # 平均法
hclust(E.dist, method="centroid") # 中心法
hclust(E.dist, method="ward.D2") # 华德法
17.对所有样本的表达矩阵进行PCA分析并且绘图,同样要添加表型信息。
pc <- prcomp(t(exprSet),scale=TRUE) #exprSet本是行为基因名,列为样本名,但t(exprSet)后就应该反过来了
pcx=data.frame(pc$x)
pcr=cbind(samples=rownames(pcx),group_list, pcx)
p=ggplot(pcr, aes(PC1, PC2))+geom_point(size=5, aes(color=group_list)) +#能最大反映样本差异性的两个成分(PC1、PC2)
geom_text(aes(label=samples),hjust=-0.1, vjust=-0.3)#label=samples可以加上样本名称
print(p)

心得:由于前面有一步将exprSet给转换成行为样本名,列为基因名了,所以这时再t(exprSet)就是错误的了,用的原始的exprSet都是行为基因名,列为样本。在这个地方捣鼓了好久。
18.根据表达矩阵及样本分组信息进行批量T检验,得到检验结果表格
exprSet=dat #从以上步骤得到的是 先是dat获得的行为基因名,列为正常顺序的样本的这样一个矩阵,给exprSet后,它俩现在一样了
group_list=as.factor(group_list)
group1 = which(group_list == levels(group_list)[1])#见下图中progress.中所示
group2 = which(group_list == levels(group_list)[2])#见下图中stable中所示
dat1 = dat[, group1]#取出分组信息为progress.的表达矩阵
dat2 = dat[, group2]##取出分组信息为stable的表达矩阵
dat = cbind(dat1, dat2)#cbind:列数形同时,横向追加。rbind:行数相同时,纵向追加
pvals = apply(exprSet, 1, function(x){ #后面导致19题出现的问题是这个地方的exprSet在之前被我给换成rbind的dat了,也就是顺序是14个progress.(11、13、14、15、16、19...),然乎再全是stable。而此时分组信息group_list就是按照最初group_list中的12、13、14、15、16、16...。由于矩阵和分组信息不是匹配的,所以不该该exprSet为cbind后的dat。那么问题是,既然用exprSet,那么为什么要有dat = cbind(dat1, dat2)这步呢?这时目前唯一不理解的啦,但是如果不用这个exprSet(行为基因名,列为正常顺序的样本),后面第20题就会做出错误的图。
t.test(as.numeric(x)~group_list)$p.value
})
p.adj = p.adjust(pvals, method = "BH")
avg_1 = rowMeans(dat1)
avg_2 = rowMeans(dat2)
log2FC = avg_2-avg_1
DEG_t.test = cbind(avg_1, avg_2, log2FC, pvals, p.adj)
DEG_t.test=DEG_t.test[order(DEG_t.test[,4]),]
DEG_t.test=as.data.frame(DEG_t.test)
# 查看t检验结果表格,包含log2FC、pvals和p.adj等,通常认为t<0.05即有统计学意义
head(DEG_t.test)
- 结果为

找到分组信息为progres.的下角标(即在dat矩阵中所处于的位置(哪一列))

找到分组信息为stable的下角标(即在dat矩阵中所处于的位置(哪一列))

- 简单罗列一下which 和 %in% 的用法,如下举例
a=c(1,3,4,5,3,2,5,6,3,2,5,6,7,5,8)
which.max(a) #which列出符合函数结果的下角标,即列出默认为TRUE的下角标
[1] 15
10:1 %in% a
[1] FALSE FALSE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
b <- which(10:1 %in% a)
b
[1] 3 4 5 6 7 8 9 10
- cbind函数用法,如下举例
> a <- matrix(1:6, 2, 3)
> print(a)
[,1] [,2] [,3] [,4]
[1,] 1 4 7 10
[2,] 2 5 8 11
[3,] 3 6 9 12
>
> b <- matrix(-1:-12, 3, 4)
> print(b)
[,1] [,2] [,3] [,4]
[1,] -1 -4 -7 -10
[2,] -2 -5 -8 -11
[3,] -3 -6 -9 -12
>
> x=cbind(a,b)
> print(x)
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8]
[1,] 1 4 7 10 -1 -4 -7 -10
[2,] 2 5 8 11 -2 -5 -8 -11
[3,] 3 6 9 12 -3 -6 -9 -12
19.使用limma包对表达矩阵及样本分组信息进行差异分析,得到差异分析表格,重点看logFC和P值,画个火山图(就是logFC和-log10(P值)的散点图。
下面代码块中的dat是18题得到的dat = cbind(dat1, dat2)的dat,把它赋值给exprSet。这就是后面为什么出现的火山图的差异表达基因和其他人不同的原因了
#exprSet=dat #这一步也不该加,因为此时的dat是18题中cbind后的dat,而我之前加了,加了后矩阵和分组信息又是不对应的了,见下图中错误的
exprSet[1:4,1:4]
# DEG by limma
suppressMessages(library(limma))
design <- model.matrix(~0+factor(group_list))
colnames(design)=levels(factor(group_list))
rownames(design)=colnames(exprSet)
design #注意此步的design图片与下张design图片是不同的,下面的exprSet才是正确的
contrast.matrix<-makeContrasts(paste0(unique(group_list),collapse = "-"),levels = design)
contrast.matrix<-makeContrasts("progres.-stable",levels = design)#关于makeContrasts下面有个示例
contrast.matrix
##这个矩阵声明,我们要把progres.组跟stable进行差异分析比较
##step1
fit <- lmFit(exprSet,design)
##step2
fit2 <- contrasts.fit(fit, contrast.matrix)
fit2 <- eBayes(fit2)
##eBayes() with trend=TRUE
##step3
tempOutput = topTable(fit2, coef=1, n=Inf)
nrDEG = na.omit(tempOutput)
#write.csv(nrDEG2,"limma_notrend.results.csv",quote = F)
head(nrDEG)
#下面这些代码是调整好的,基本可以不用替换的呢
DEG=nrDEG
logFC_cutoff <- with(DEG,mean(abs(logFC)) + 2*sd(abs(logFC)) )
DEG$result = as.factor(ifelse(DEG$P.Value < 0.05 & abs(DEG$logFC) > logFC_cutoff,
ifelse(DEG$logFC > logFC_cutoff ,'UP','DOWN'),'NOT'))
this_tile <- paste0('Cutoff for logFC is ',round(logFC_cutoff,3), #round保留小数位数
'\nThe number of up gene is ',nrow(DEG[DEG$result =='UP',]) ,
'\nThe number of down gene is ',nrow(DEG[DEG$result =='DOWN',])
)
ggplot(data=DEG, aes(x=logFC, y=-log10(P.Value), color=result)) +
geom_point(alpha=0.4, size=1.75) +
theme_set(theme_set(theme_bw(base_size=20)))+
xlab("log2 fold change") + ylab("-log10 p-value") +
ggtitle( this_tile ) + theme(plot.title = element_text(size=15,hjust = 0.5))+
scale_colour_manual(values = c('blue','black','red'))

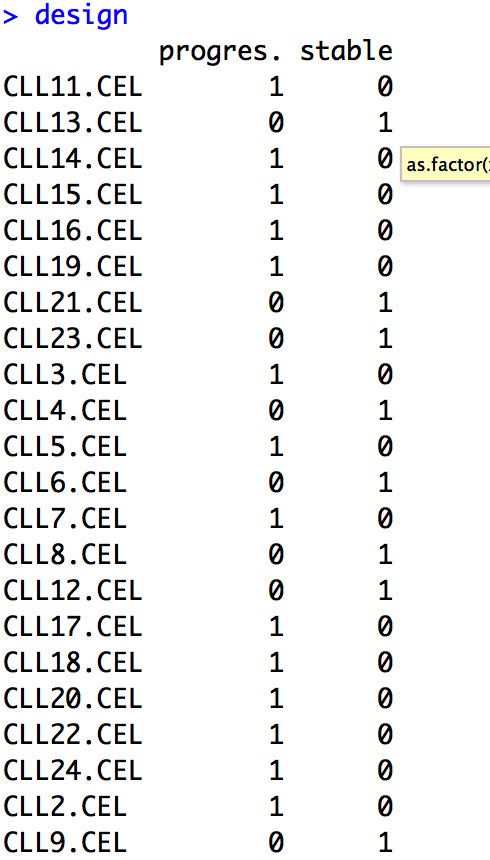

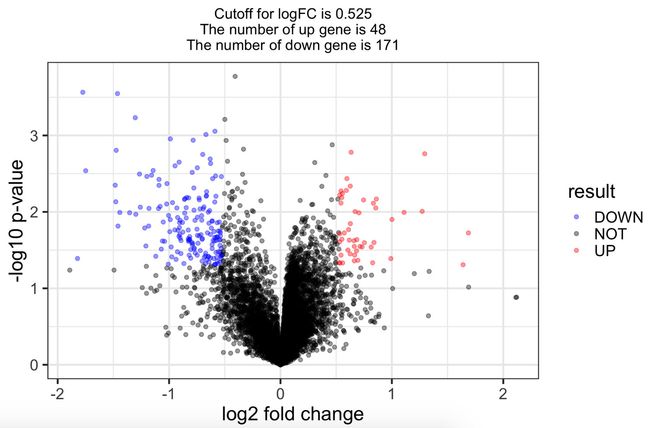
- 插播插曲
接下来下面3张图片的代码与上面的代码相同,只不过第一步中exprSet=dat的dat是未经过第18题中的dat = cbind(dat1, dat2)的dat,而是第10题中的dat,就是说,用的exprSet就是原始从第2题中提取出的表达矩阵,经过同ids对应好以后的exprSet。因此样本信息也是按照没有修改过的顺序11、12、13、14、15来的,同时design信息即样本和分组信息的对应是正确的,因为这个里面的exprSet是最初的,其实group_list一直都没有变过,变的是矩阵,即上面的dat是dat1和dat2组合在一起的,它的样本顺序是改变了的,是dat1全部是progres.,而dat2全部是stable。但是我们并没有操作group_list,group_list是最开始第2题中从pData中提取出来的。所以在上面图中design的样本和分组信息是错误对应的。因此上面最后画出的火山图也是错误的。总结来说,这里面的分组信息和矩阵exprSet是独立来的,可以class(exprSet)可以得到matrix,而不是data frame,所以在把dat分为progres.和stable后在cbind这两组后,并不能让group_list跟着一起分组。
#下面生成图的代码同上面的代码一样,只不过第一步中exprSet=dat的dat是未经过第18题中的dat = cbind(dat1, dat2)的dat,而是第10题中的dat,即行为基因名字 ,列为同exprSet中一样的顺序。

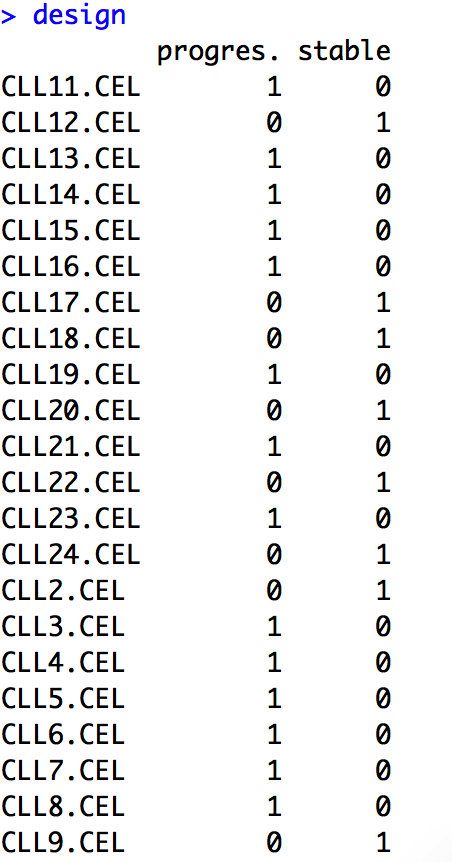


- 代码中设计的函数的一些简单示例,如下
- makeContrasts函数用法,如下举例
x <- c("B-A","C-B","C-A")
makeContrasts(contrasts=x,levels=c("A","B","C"))
Contrasts
Levels B - A C - B C - A
A -1 0 -1
B 1 -1 0
C 0 1 1
#相当于下面这个,输出结果是一样的
makeContrasts(B-A,C-B,C-A,levels=c("A","B","C"))
- unique、duplicated函数用法,如下举例
#unique主要是返回一个把重复元素或行给删除的向量、数据框或数组
> rt
年 月 公司名 利率
1 2000 1 A a
2 2000 1 A a
3 2001 2 A b
4 2001 3 A c
5 2000 1 B d
6 2000 2 B e
7 2000 2 B e
> unique(rt)
年 月 公司名 利率
1 2000 1 A a
3 2001 2 A b
4 2001 3 A c
5 2000 1 B d
6 2000 2 B e
> unique(rt,fromLast=TRUE)
年 月 公司名 利率
2 2000 1 A a
3 2001 2 A b
4 2001 3 A c
5 2000 1 B d
7 2000 2 B e
以上是根据你的数据得到的,R中默认的是fromLast=FALSE,即若样本点重复出现,则取首次出现的;
否则取最后一次出现的。
#duplicated主要是判定向量或数据框中的元素是否重复,它返回一个元素(行)是不是重复的逻辑向量
> data.set
Ensembl.Gene.ID Gene.Biotype Chromosome.Name Gene.Start..bp. Gene.End..bp.
1 ENSG00000236666 antisense 22 16274560 16278602
2 ENSG00000236666 antisense 22 16274560 16278602
3 ENSG00000234381 pseudogene 22 16333633 16342783
4 ENSG00000234381 pseudogene 22 16333633 16342783
5 ENSG00000234381 pseudogene 22 16333633 16342783
6 ENSG00000234381 pseudogene 22 16333633 16342783
7 ENSG00000234381 pseudogene 22 16333633 16342783
8 ENSG00000234381 pseudogene 22 16333633 16342783
9 ENSG00000234381 pseudogene 22 16333633 16342783
10 ENSG00000224435 pseudogene 22 16345912 16355362
#构建一个布尔向量,索引
> index<-duplicated(data.set$Ensembl.Gene.ID)
> index
[1] FALSE TRUE FALSE TRUE TRUE TRUE TRUE TRUE TRUE FALSE
#筛选数据
> data.set2<-data.set[!index,] #选中了非重复的数据
- paste和paste0函数举例
#R连接函数paste和paste0
#paste()与paste0()不仅可以连接多个字符串,还可以将对象自动转换为字符串再相连,另外还能处理向量
paste("fitbit", month, ".jpg", sep="")
#这个函数的特殊地方在于默认的分隔符是空格,所以必须指定sep="",这样如果month=10时,就会生成fitbit10.jpg这样的字符串
#要生成12个月的fitbit文件名
paste("fitbit", 1:12, ".jpg", sep = "")
[1] "fitbit1.jpg" "fitbit2.jpg" "fitbit3.jpg" "fitbit4.jpg" "fitbit5.jpg" "fitbit6.jpg" "fitbit7.jpg"
[8] "fitbit8.jpg" "fitbit9.jpg" "fitbit10.jpg" "fitbit11.jpg" "fitbit12.jpg"
#可以看出参数里面有向量时的捉对拼接的效果,如果某个向量较短,就自动补齐
a <- c("甲","乙","丙","丁","戊","己","庚","辛","壬","癸")
b <- c("子","丑","寅","卯","辰","巳","午","未","申","酉","戌","亥")
paste0(a, b)
[1] "甲子" "乙丑" "丙寅" "丁卯" "戊辰" "己巳" "庚午" "辛未" "壬申" "癸酉" "甲戌" "乙亥"
#collapse是合并成一个字符串时的分隔符
paste("fitbit", 1:3, ".jpg", sep = "", collapse = "; ")
[1] "fitbit1.jpg; fitbit2.jpg; fitbit3.jpg"
20.对T检验结果的P值和limma包差异分析的P值画散点图,看看哪些基因相差很大?
head(nrDEG)
head(DEG_t.test)
DEG_t.test=DEG_t.test[rownames(nrDEG),]
plot(DEG_t.test[,3],nrDEG[,1]) ## 可以看到logFC是相反的
plot(DEG_t.test[,4],nrDEG[,4]) # 可以看到使用limma包和t.test本身的p值差异尚可接受
plot(-log10(DEG_t.test[,4]),-log10(nrDEG[,4]))

- 最后画出的三图是这样的
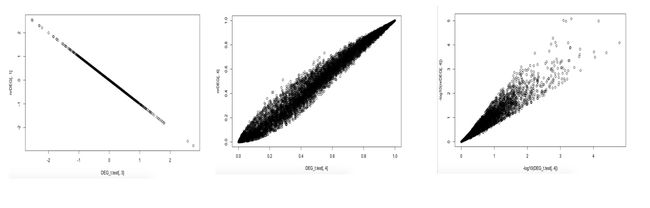
- 不然,按照我原来的思路,第二张图的画风是这样的,所以说老大一直说的,你要时刻知道你现在操作的
对象是什么,就是赤裸裸的真理啊,不然会激起密集恐惧症的,哈哈。

最后,还有个疑问,那就是第18题中的疑问,当时看别人的代码,把exprSet给改成了cbind后的代码,后来向老大要来老大代码,其实就在R-20题里呢,没有找到。发现那个地方是和老大一样的,所以那18题里的第8行代码中用一开始的未经cbind的正常顺序样本的矩阵,那为什么要有dat = cbind(dat1, dat2)这步呢?
最后再仔细想想老大的代码,截图如下:

我想这个cbind后的dat就是合起来看一下,了解一下cbind的用法,而我确实也是又去查了cbind的用法(想知道它是怎么用的,和merge的区别,merge是用by的,通过相同的一列来叠加(如by probe_id)而cbind是行数相同横向追加),从图中粉色圈出的地方可以看的其实后面用的dat1和dat2。所以我觉得是就可以这样解释了!疑问解决了。开心。
总结,对象很重要,你要时刻知道你现在面对的对象是谁!