table_annovar.pl
运行该程序时,会根据代码需要拆成几个步骤运行,如先用convert2annovar将vcf转为avinput文件,再根据protocol中的内容拆成几项annotate_variation.pl任务。
- Example:
/mnt/fvg01vol8/software/biosoft/annovar/table_annovar.pl /mnt/fvg01vol7/project/180601_E00599_0078_AH52NWCCXY/MCGHUM1805002E/result//vari/XWJ_180428/XWJ_180428.vcf /mnt/fvg01vol8/database/humandb/annovardb -out /mnt/fvg01vol7/project/180601_E00599_0078_AH52NWCCXY/MCGHUM1805002E/result//Annotation/XWJ_180428/XWJ_180428.vari -buildver hg19 -remove -protocol refGene,cytoBand,genomicSuperDups,esp6500siv2_all,1000g2015aug_all,1000g2015aug_afr,1000g2015aug_eas,1000g2015aug_eur,snp138,ljb26_all -operation g,r,r,f,f,f,f,f,f,f -nastring . -vcfinput
- 常见参数说明:
Usage:
table_annovar.pl [arguments]
optional arguments:
--protocol comma-delimited string specifying database protocol
--operation comma-delimited string specifying type of operation
--outfile output file name prefix
--buildver genome build version (default: hg18)
--remove remove all temporary files
--nastring string to display when a score is not available (default: null)
--csvout generate comma-delimited CSV file (default: tab-delimited txt file)
--gff3dbfile specify comma-delimited GFF3 files
--vcfinput specify that input is in VCF format and output will be in VCF format
Input data prepare
The convert2annovar.pl script can convert other "genotype calling" format into ANNOVAR format. Currently, the program can handle Samtools genotype-calling pileup format, Illumina CASAVA format, SOLiD GFF genotype-calling format, Complete Genomics variant format, SOAPsnp format, MAQ format and VCF format. Additionally, the program can generate ANNOVAR input files from a list of dbSNP identifiers, or from transcript identifiers, or from a genomic region.
- 常见参数说明:
USAGE: convert2annovar.pl [arguments]
--format
the format of the input files. Currently supported formats
include pileup, cg, cgmastervar, gff3-solid, soap, maq, casava,
vcf4, vcf4old, rsid. In August 2013, the VCF file processing
subroutine is changed (multiple samples in VCF file can be
processed in genotype-aware manner), but users can use vcf4old
to have identical results as the old behavior. (输入文件格式,常
用的VCF4)
--outfile
specify the output file name. By default, output is written to
STDOUT. (输出文件,否则就打印到屏幕上,或可采用'>'重定向到文件)
--allsample
for multi-sample **VCF4** file, the --allsample argument will
process all samples in the file and generate separate output
files for each sample. By default, only the first sample in VCF4
file will be processed. (每个样本生成一个avinput文件)
--withzyg
for VCF4 format, print out zygosity information, coverage
information and genotype quality information when -includeinfo
is used. By default, these information are printed out if
-includeinfo is not used. (输出纯杂合信息、覆盖度、基因型质量)
--snpqual
quality score threshold in the pileup file, such that variant
calls with lower quality scores will not be printed out in the
output file.(PILEUP文件时所采用的的过滤条件)
- avinput格式:
- chromosome
- start
- end
- reference allele
- alternative allele
- annotation [OPTION]
eg.
20 1110696 1110696 A G het 67 6
*The 3 extra columns are zygosity status, genotype quality and read depth.
-
ps:
- In some cases, users may want to specify only positions but not the actual nucleotides. In that case, "0" can be used to fill in the 4th and 5th column.
- If ANNOVAR encounters an invalid input line, it will write the invalid line into a file called {outfile}.invalid_input
Gene-based annotation
- 数据库的下载和准备:
Before working on gene-based annotation, a gene definition file and associated FASTA file must be downloaded into a directory if they are not already downloaded.
获取Gene Definition文件:
annotate_variation.pl -downdb -buildver hg19 -webfrom annovar refGene humandb/
humandb/为本地数据库存放位置,RefGene参考RefGene, UCSC说明
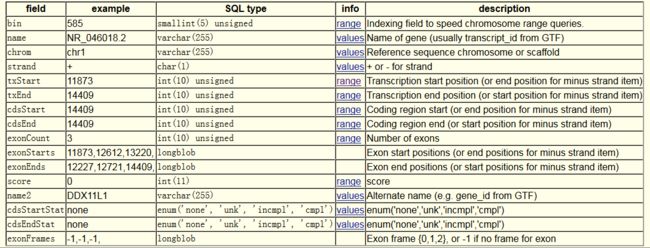
For other gene definition systems (such as GENCODE, CCDS) or for other species (such as mouse/fly/worm/yeast), the user needs to build the FASTA file yourself.
- 注释过程:
参数说明:
Arguments to download databases or perform annotations
--downdb download annotation database
--geneanno annotate variants by gene-based annotation (infer functional consequence on genes)
--regionanno annotate variants by region-based annotation (find overlapped regions in database)
--filter annotate variants by filter-based annotation (find identical variants in database)
Arguments to control input and output
--outfile output file prefix
--webfrom specify the source of database (ucsc or annovar or URL) (downdb operation)
--dbtype specify database type
--buildver specify genome build version (default: hg18 for human)
基础语法refgene注释:其中-geneanno,-dbtype refGene为默认参数,注意RefGene不含Mt信息
annotate_variation.pl -out ex1 -build hg19 example/ex1.avinput humandb/
注释后生成两个结果文件: ex1.refGene.exonic_variant_function和ex1.refGene.variant_function, 一个为变异信息,一个为外显子区域变化情况
UCSC注释:The transcript name look like uc002eg1.1, etc.
annotate_variation.pl -out ex1 -build hg19 example/ex1.avinput humandb/ -dbtype knownGene
Ensemble注释:ensemblToGeneName.txt.gz can translate Ensembl identifiers to gene synonym.
annotate_variation.pl -out ex1 -build hg19 ex1.hg19.avinput humandb/ -dbtype ensGene
Technical Notes: Technically, the RefSeq Gene and UCSC Gene are transcript-based gene definitions. They built gene model based on transcript data, and then map the gene model back to human genomes. In comparison, Ensemble Gene and Gencode Gene are assembly-based gene definitions that attempt to build gene model directly from reference human genome. They came from different angles, trying to do the same thing: define genes in human genome.
- 其他物种:
The GFF3 or GTF file downloaded from Ensembl or compiled by the user need to be converted to the GenePred format performed by gff3ToGenePred or gtfToGenePred.
- Please decompress both files (GTF file and the genome FASTA file for this plant):
解压GTF(GFF)文件和fasta文件
gunzip Arabidopsis_thaliana.TAIR10.27.dna.genome.fa.gz
gunzip Arabidopsis_thaliana.TAIR10.27.gtf.gz
- Please use the
gtfToGenePredtool to convert the GTF file to GenePred file:
用软件gtfToGenePred将GTF文件转为refGene格式
gtfToGenePred -genePredExt Arabidopsis_thaliana.TAIR10.27.gtf AT_refGene.txt
- Please generate a transcript FASTA file with our provided script:
用annovar的retrieve_seq_from_fasta.pl软件生成转录组序列文件
perl retrieve_seq_from_fasta.pl --format refGene --seqfile Arabidopsis_thaliana.TAIR10.27.dna.genome.fa AT_refGene.txt --out AT_refGeneMrna.fa
After this step, the annotation database files needed for gene-based annotation are ready. Now you can annotate a given VCF file. Please note that the --buildver argument should be set to AT.
Region Based Annotation
Filter-based annotation是对变异位点的注释,而Region-based annotationz主要针对的是那段区域。可采用的数据库:UCSC数据库、BED文件、GFF文件。
Filter-based annotation looks exact matches between a query variant and a record in a database; two items are identical only if they have identical chromosome, start position, end position, ref allele and alaternative allele. Region-based annotation looks for over lap of a query variant with a region (this region could be a single position) in a database, and it does not care about exact match of positions, and it does not care about nucleotide identity at all.
- UCSC下载文件注释
UCSC相关数据可使用Annovar自带软件下载
annotate_variation.pl -buildver hg19 -downdb targetScanS ~
注释:
annotate_variation.pl -region ./test.avinput humandb/ -buildver hg19 -dbtype targetScanS -out test
- GFF注释
GFF3格式说明,注释之后生成一个ex1.hg19_gff3文件,其中"Name="之后的内容即为GFF文件所对应的ID号
annotate_variation.pl -regionanno -dbtype gff3 -gff3dbfile hg18_example_db_gff3.txt ex1.hg18.avinput humandb/ -out ex1
- BED文件注释
annotate_variation.pl ex1.hg18.avinput humandb/ -bedfile hg18_SureSelect_All_Exon_G3362_with_names.bed -dbtype bed -regionanno -out ex1
参考网址:
- ANNOVAR注释软件(CSDN)
- ANNOVAR Document
测试记录:
- 2018-06-06测试芒草基因组注释:
/mnt/fvg01vol8/software/biosoft/gff3ToGenePred /mnt/fvg01vol8/database/Plants/Miscanthus_sinensis/v7.1/annotation/Msinensis_497_v7.1.gene_exons.gff3 /mnt/fvg01vol8/database/Plants/Miscanthus_sinensis/v7.1/annotation/Mis_refGene.txt
报错信息:
/mnt/fvg01vol8/database/Plants/Miscanthus_sinensis/v7.1/annotation/Msinensis_497_v7.1.gene_exons.gff3:2: invalid meta line: ##annot-version v7.1
/mnt/fvg01vol8/database/Plants/Miscanthus_sinensis/v7.1/annotation/Msinensis_497_v7.1.gene_exons.gff3:3: expected "##species NCBI_Taxonomy_URI", got "##species Miscanthus sinensis"
GFF3: 2 parser errors
解决方法:
删除2、3行信息,但保留第一行##gff-version 3
不保留第一行将会有如下报错:
/mnt/fvg01vol8/database/Plants/Miscanthus_sinensis/v7.1/annotation/Msinensis_497_v7.1.gene.gff3:1: invalid GFF3 header
Can't find annotation record "Misin01G000100.v7.1" referenced by "Misin01G000100.1.v7.1" Parent attribute
GFF3: 2 parser errors
ERROR:
程序完整运行,未出现报错信息,但是注释出来的全是
intergenic,且Gene.refGene部分显示为NONE;NONE
解决方法:
VCF中chromosome一栏所显示的染色体号仅有数字1、2...19,而注释的GFF文件中不同染色体表示为Chr01...,修改VCF文件后,可正确注释SNP位点。
其他注释相关软件
snpEff