BETA 网址: http://cistrome.dfci.harvard.edu/BETA/tutorial.html
功能介绍
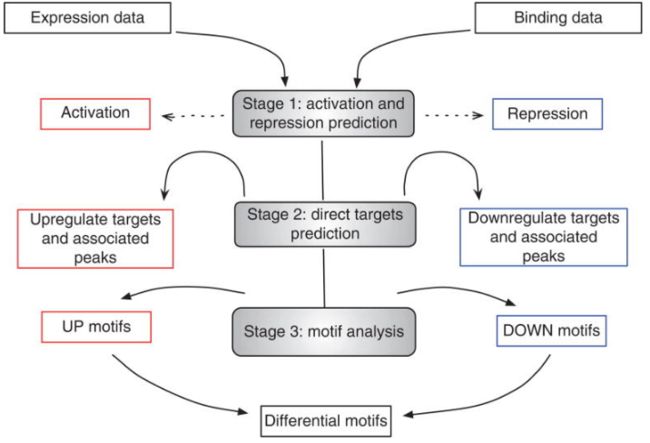
数据准备:
- peak bed file
# chr str end
$head mep_ebDfpeak_deseq2_sig.bed
chr8 12050808 12052743
chr9 66492999 66494729
chr4 125632449 125634758
chr4 10182030 10183501
- df expression file
# symbol log2FC pvalue
$head mep_eb_dfexpre
MELK 6.23721078699297 5.47784458574045e-218
RAD51AP1 4.17541170218602 2.86774188064759e-217
HMGN5 5.98876520905166 7.49359285750074e-216
ZWILCH 3.74468061674801 2.42032701371558e-204
HNRNPA2B1 2.76252025379004 3.05116228100538e-190
HLA-DRA -5.19413815362273 2.27680965999626e-188
软件下载安装
wget http://cistrome.dfci.harvard.edu/BETA/src/BETA_1.0.7.zip
unzip BETA_1.0.7.zip
cd BETA_1.0.7/
sudo python setup.py install
使用
BETA Basic
BETA Basic 预测调控因子的功能(激活或抑制)和靶基因
BETA basic –p 3656_peaks.bed –e AR_expr.xls –k LIM –g hg19 --da500 –n basic
参数:
-p : peak bed file (必需参数)
-e:df expression file (必需参数)
-k:表达量文件格式是limma的标准格式(根据实际情况修改)
-g:基因组文件,如hg19,hg38,mm10,mm9
-n:输出文件前缀,BETA默认是NA
-da:选择显著性变化(上调和下调的)表达数据(根据实际情况修改)
其他格式(不是limma和cuffdif标准格式)的差异表达文件格式:
- 需设置参数
--info, geneID如果是smbol需要设置--gname2
BETA Plus
BETA Plus 预测调控因子的功能(激活或抑制)和靶基因,以及靶向区域的motif分析
BETA plus –p 3656_peaks.bed –e AR_expr.xls –k LIM –g hg19 --gs hg19.fa --bl
参数:
--gs : motif分析需要的参数, 基因组fasta格式
--bl :可选参数,当考虑boundary(如CTCF)时需要的参数
BETA Minus
BETA Minus 只根据peak bed数据基于调控潜能值预测靶基因
BETA minus -p 3656_peaks.bed --bl -g hg19
其他可选参数
-n:结果文件的名字
-o:输出文件路径
--gname2:geneID如果是smbol需要设置--gname2
--info:其他格式(不是limma和cuffdif标准格式)的差异表达文件格式:需设置参数--info,
--pn:peak 数目,默认是10,000
-d DISTANCE,--distance 距离TSS的距离. DEFAULT=100000 (100kb)
--df DIFF_FDR : 输入0-1的数值,表示统计的阈值,如FDR等,默认是1.
--da DIFF_AMOUNT; 取显著表达的基因,0-1表示比例,大于1表示个数。如2000,表示去top 2000个上调和下调的差异基因;0.5表示取50%上调和下调的基因,默认是0.5。
-c CUTOFF, --cutoff: 0~1的一个阈值。默认是1e-3。
-r REFERENCE, --reference: 当参考基因组不是hg19,或mm9时使用此参数。
--bl BOUNDARY :LIMIT Boolean Value. Whether or not use CTCF boundary to get a peak’s associated gene, DEFAULT=FALSE
-bf BOUNDARYFILE : Some BED format boundary file, use this only when You set --bl and the genome is neither hg19 nor mm9
相关资料
文章Wang, S., Sun, H., Ma, J., Zang, C., Wang, C., Wang, J., ... & Liu, X. S. (2013). Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nature protocols, 8(12), 2502-2515.
PMID: 24263090
网址: http://cistrome.org/BETA/index.html
使用代码
/software/biosoft/software/python/python2.7/bin/BETA plus -P mep_ebDfpeak_deseq2_sig.bed --info -e mep_eb_dfexpre --gname2 -g hg 19 --gs hg19.fa --bl
几点疑问
BETA最初的设计主要是用于ChIP-Seq和RNA-seq的整合分析,最终可以预测特定一个转录因子的靶基因,以及这些基因是激活还是抑制的功能。但是对于ATAC-seq,它得到的信号整个基因组范围转录因子的信号,那么与转录组整合分析得到的结果怎么解释?只能知道一些基因的功能是激活的或抑制的,但是并不能知道这些基因是哪些转录因子的靶基因。所以BETA是否适合ATAC-seq和与RNA-seq的整合分析?如果可以,整合分析的结果能得到哪些有价值的信息?