1 比对的是:使用idba_ud拼接的AER314-4raw_data基因组与转录组数据。
2 bowtie2做index(bowtie2使用conda安装)
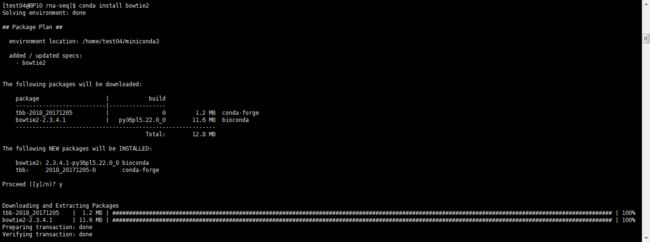
建索引:bowtie2-build AER314-4_scaffold.fa AER314-4_scaffold.fa
3 reads mapping到参考基因组——tophat2软件:基于bowtie2(tophat安装见软件安装)
命令:tophat2 -p 12 -o AER314-4_output /home/test04/lyr/rna-seq/02align_out/AER314-4_scaffold.fa /home/test04/lyr/rna-seq/01data/YSH-qurRNA-42-314-4_L001_R1.fastq /home/test04/lyr/rna-seq/01data/YSH-qurRNA-42-314-4_L001_R2.fastq
4 然后就很顺利的跑出来结果了
使用公司服务器,12个线程,大概五个小时就跑完啦。
5 cufflink
[ Cufflinks输出结果
cufflinks的输入文件是sam或bam格式。并且sam或bam格式的文件必须排好序。(The SAM file supplied to Cufflinksmustbe sorted by reference position.)Tophat的输出结果sam或bam已经排好了序。针对其他的未排序的sam或bam文件采用如下排序方式:
sort -k 3,3 -k 4,4n hits.sam > hits.sam.sorted
1. transcripts.gtf
该文件包含Cufflinks的组装结果isoforms。前7列为标准的GTF格式,最后一列为attributes。其每一列的意义:
列数 列的名称 例子 描述
1 序列名 chrX 染色体或contig名; 2 来源 Cufflinks 产生该文件的程序名; 3 类型 exon 记录的类型,一般是transcript或exon; 4 起始 1 1-base的值; 5 结束 1000 结束位置; 6 得分 1000 ; 7 链 + Cufflinks猜测isoform来自参考序列的那一条链,一般是'+','-'或'.';8 frame . Cufflinks不去预测起始或终止密码子框的位置; 9 attributes ... 详见下
每一个GTF记录包含如下attributes:
Attribute 例子 描述
gene_idCUFF.1Cufflinks的gene id;transcript_idCUFF.1.1 Cufflinks的转录子 id; FPKM 101.267 isoform水平上的丰度,FragmentsPerKilobase of exon model perMillion mapped fragments; frac 0.7647 保留着的一项,忽略即可,以后可能会取消这个;conf_lo 0.07 isoform丰度的95%置信区间的下边界,即 下边界值 = FPKM * ( 1.0 - conf_lo );conf_hi 0.1102 isoform丰度的95%置信区间的上边界,即 上边界值 = FPKM * ( 1.0 + conf_hi ); cov 100.765 计算整个transcript上read的覆盖度;full_read_support yes 当使用 RABT assembly 时,该选项报告所有的introns和exons是否完全被reads所覆盖
2. ispforms.fpkm_tracking
isoforms(可以理解为gene的各个外显子)的fpkm计算结果
3. genes.fpkm_tracking
gene的fpkm计算结果Cuffmerge简介
Cuffmerge将各个Cufflinks生成的transcripts.gtf文件融合称为一个更加全面的transcripts注释结果文件merged.gtf。以利于用Cuffdiff来分析基因差异表达。
2. 使用方法
$ cuffmerge [options]*
输入文件为一个文本文件,是包含着GTF文件路径的list。常用例子:
$ cuffmerge -o ./merged_asm -p 8 assembly_list.txt
3. 使用参数
-h | --help
-o default: ./merged_asm
将结果输出至该文件夹。
-g | --ref-gtf将该reference GTF一起融合到最终结果中。
-p | --num-threads defautl: 1
使用的CPU线程数
-s | --ref-sequence /该参数指向基因组DNA序列。如果是一个文件夹,则每个contig则是一个fasta文件;如果是一个fasta文件,则所有的contigs都需要在里面。Cuffmerge将使用该ref-sequence来帮助对transfrags分类,并排除repeats。比如transcripts包含一些小写碱基的将归类到repeats. ]
cufflinks:
<1>命令:cufflinks -p 4 -o test_cuff /home/andengdi/lyr/rna-seq/02-align_out/test_output/accepted_hits.bam
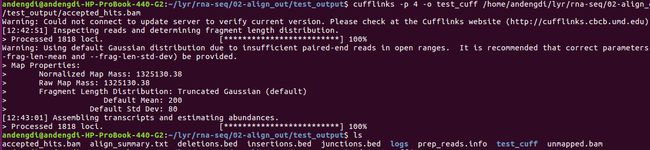
流程及结果
5 用相同的方法将其他两个样本跑一遍。