计算抗疫

 技术抗疫的典型。 作者
|Decode
技术抗疫的典型。 作者
|Decode
邮箱 |[email protected]
每一场战役,都有幕后英雄。在抗击新型冠状病毒肺炎(以下简称新冠肺炎)中,“计算”是不容易被看见的力量。
由于生物信息学和计算生物学的发展,现代生命科学相关研究已经离不开强大的计算能力。在一场传染病中找出“敌人”是谁、弄清“敌人”长什么样以及研制药物,每个环节都有海量需要计算的生物大数据。
如果只采用普通计算机,耗时要数周甚至数月,而高性能计算系统只需几个小时甚至几十分钟就能完成。

揪出敌人
准确揪出病原,是对抗疫情关键的第一步。生产检测试剂、临床用药和研发疫苗,都以找准病原体为基础。
据《中国青年报》报道,2003 年中国爆发 SARS 疫情,北京一位疾控专家通过电子显微镜,就判断病原为衣原体。衣原体导致的肺炎流行概率低,死亡率不高,也有特效药可治。2 月 18 日,权威媒体报道了这一消息后,给了公众“非典不可怕”错觉。
“SARS 最惨痛的教训就是长时间无法确认病原。” 中国工程院院士、国家卫健委高级别专家组成员李兰娟,在接受经济日报采访时说,“那个时候我国识别病原的能力相对薄弱,在疫情发生半年后才由美国科学家确认病原。”
2019 年底新冠肺炎疫情来袭,情况已经完全不一样。
据财新报道,2019 年 12 月 24 日,武汉市中心医院对一位老年病人进行了采样,样本被送到广州微远基因进行测序。几日前,他因发烧前往该医院就诊,被怀疑是“社区获得性肺炎”。微远基因是基因科技与精准医疗方向的企业,有快讯诊断病原技术。
2020 年 1 月 27 日发表在《中华医学杂志(英文版)》上的一篇论文显示,这位老人是较早进行基因测序的临床样本。
12 月 27 日,微远基因就组装出接近完整的新型病毒基因组序列,并且向上述医院和疾控部门反馈了测序结果,“是一种新的冠状病毒”。
12 月 30 日,中科院武汉病毒所收到武汉市金银潭医院送来的样品。2020 年 1 月 2 日,该所就确定了 2019 新冠病毒的全基因组序列。
从几个月到几天,中国揪出病原时间大大缩短。这背后,多家实验室都用到了一种关键技术——mNGS。
mNGS 全称 metagenomics next generation sequencing(宏基因组新一代测序技术),是近几年基因技术的热点方向。2019 年 11 月的北京鼠疫疫情,也是通过 mNGS 检测出来的。

这项技术涉及两个概念:宏基因组和新一代测序技术。前者是目前研究环境微生物的主流思路,过去准入门槛很高,测序和信息分析价格昂贵,因此应用一直比较有限。
直到新一代测序技术出现,才将宏基因组学推向了前台。新一代测序技术又称为“高通量测序”和“深度测序”,一次能并行对几百万条基因进行测序,大大缩短了时间,并且让成本呈几何级数地下降。
在这次新冠肺炎疫情中,基于实时荧光定量 PCR(RT-PCR)的核酸检测和 mNGS,都是最早用于确诊新冠病毒感染的手段。
前者操作简单、成本低且速度快,但准确率不高;后者一次检测便能排查所有已知病原体,可以防止病毒变异而导致的漏检,但相对而言操作复杂、检测时间较长。因此,一线医生通常联合使用两种手段。
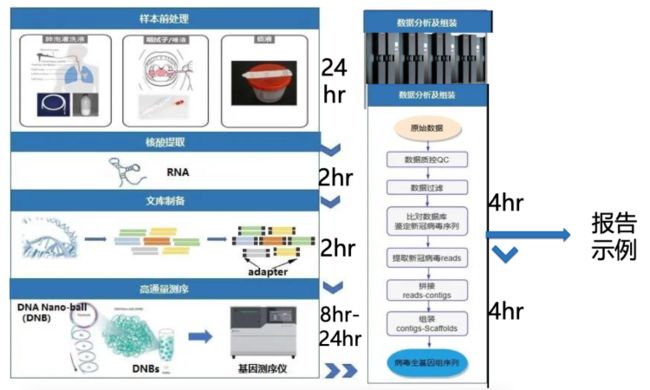
2020 年 3 月的一次在线分享中,浪潮生命科学行业经理成嵩婷,介绍了某疾控中心操作 mNGS 的完整流程:“前期,运送加上样本收集需要 24 小时,接着做核酸提取和文库制备。制备完了测序开始,正常是 24 小时。如果着急的话,可以用一些测序仪降到 8 个小时。”
 (图片来自浪潮和华大智造)
(图片来自浪潮和华大智造)
测序产生了海量数据,通常可达 TB(太字节,1TB=1024GB)级别。比如,华大智造于 2019 年交付商用的测序仪 DNBSEQ-T7,日产出数据最高为 6 TB。这家公司与华大基因同属华大集团旗下,是 2013 年由该集团的测序仪板块独立拆分而来。
拿到基因组数据后,还要进行生物信息分析。2018 年发表于《生物产业技术杂志》的一篇文章指出,高通量测序所生成的原始数据,并不能反映任何有价值的信息,必须通过专业分析和解读。
也就是说,新一代测序技术只解决了获取基因数据的效率问题,高效分析这些数据的任务,落到了计算模块上。
“(生物信息分析)包括质控、过滤和比对基因组。”成嵩婷说,“比对基因组这一步出来就是报告了,这个报告可以直接宣告取样的这个人,有没有新冠肺炎。”
基因组比对的基准,是已经公开的新冠病毒序列。在全球共享流感数据倡议组织(简称 GISAID)平台和病毒学网站(virological.org)上,有十多个国家科学家共享的新冠病毒全基因组序列。
在比对环节之后,如果还想进一步研究,可以再提取新冠病毒序列,然后做拼接和组装,生成病毒全基因序列。由于新一代测序技术局限性,测序机输出基因序列都是一小段一小段的。根据每段的重叠区域拼接起来,才能得到完整的全基因序列。
整个过程就好比“给你一座堆满了拼图片的大山,让你拼一幅图出来”。这里面计算是海量的,并且重叠区域使得基因数据膨胀了好几倍,让处理过程成为了计算密集型操作。
 (图片来自华为云社区)
(图片来自华为云社区)
通常,基因测序公司会通过引进高性能计算机或服务器集群,来对应大规模的计算存储挑战。比如,英特尔和联想合作为华大基因提供了一个大型高性能计算集群。
这个计算集群整合了联想的 GOAST 技术(基因组优化和可扩展性工具)。据联想介绍,这是首个获得英特尔精选解决方案验证的基因组分析工具,能提供 27-40 倍的性能优化。
不过,本地的计算集群建设和维护成本巨大,缺乏灵活性。云计算以其弹性扩展和按需付费的优势,正成为越来越多基因测序厂商的选择。AWS 和阿里云等服务商,都在生物信息计算领域耕耘了好几年。2 月,浙江省上线的自动化全基因组检测分析平台,就采用了阿里云的计算资源。
在这次新冠肺炎疫情中,阿里云也把基因计算云服务 AGS(Alibaba Genomics Service)免费开放给了科研机构。这项服务最快 60 秒就能完成病毒基因比对工作——通常这一过程需要 30 分钟。
“AGS 主要对 IO(磁盘读写)效率进行了改进,同时借助云上弹性调度优化,计算并行度大幅提升。”阿里云高级技术专家李鹏告诉 PingWest 品玩,“处理一组宏基因组数据量为 22M reads(读长)的测序数据,使用 1 张 GPU 显卡来加速,就能达到理想并行效果。”

摸透敌人
测出新冠病毒全基因组序列,意味着知道了对手是谁。但想弄清对手长什么样,还要进一步研究其蛋白质的三维(立体)结构。
微软研究院一篇科普文章介绍,如果把基因组序列比喻为标识一个人的身份信息,蛋白质三维结构就是身形容貌。
只有弄清楚了三维结构,才能研究病毒致病机理,进而研发药物——现代制药流程一般是,根据三维结构去筛选适合的药物化合物。
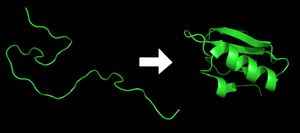
然而,弄清蛋白质三维结构不是一件简单的事情。这个“蛋白质折叠问题”,是 21 世纪生物物理学的重要课题。
蛋白质结构共分为四级,三维结构对应第三级结构,由一级结构氨基酸序列折叠而来。
 (蛋白质折叠前后/图片来自维基百科)
(蛋白质折叠前后/图片来自维基百科)
据清华大学周培源应用数学研究中心介绍,虽然蛋白质可在短时间中从一级结构折叠至立体结构,研究者却无法在短时间中,从氨基酸序列计算出蛋白质结构,甚至无法得到准确三维结构。
因此,研究蛋白质折叠过程,可以说是破译 “第二遗传密码”—— 折叠密码(folding code)的过程。
要弄清病毒的蛋白质结构,目前有两种主要方法:一种是遵循实验学,利用成像技术来构建;另一种则是基于统计学和人工智能进行预测。
实验学方法常用的技术有冷冻电镜(全称冷冻电子显微镜技术)、X 射线晶体学和核磁共振。原先,X 射线衍射是解析蛋白质结构最常用的技术,但要事先获得晶体。对有的蛋白来说,完成这项准备工作并不那么容易。
后来,冷冻电镜技术克服了一些自身局限性,名气和适用范围越来越大,2017 年还获得了诺贝尔化学奖。
“新冠病毒如何传染人”这个关键问题,正是在冷冻电镜帮助下得出答案的。
2020 年 2 月 20 日,《科学》杂志发表了一篇论文《预融合构象中 2019-nCoV 刺突的低温电镜结构》。
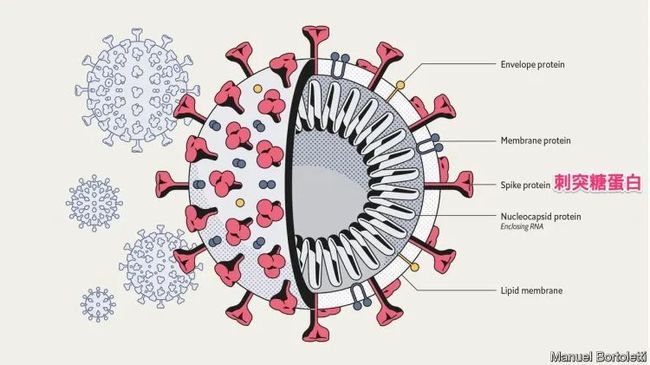
研究人员利用冷冻电镜技术,分析了新冠病毒表面 S 蛋白的结构,发现它与 SARS 病毒有着相似的感染机制。
S 蛋白全称刺突糖蛋白(spike glycoprotein),位于新冠病毒最外层,像一个个突起的 “皇冠”,通过和人体的 ACE2(宿主细胞受体血管紧张素转化酶 2)结合后传染人类。
 (图片来自于 The Economist)
(图片来自于 The Economist)
这项研究用到的冷冻电镜,原理是把样品冷冻固定住,然后在低温下用透射电镜得到二维投影图像,最后通过一系列建模和变换,转变为三维结构。
三维重构是冷冻电镜方法耗时最长的环节。“在这过程中,高性能计算能够加速三维结构重建……冠状病毒整体分子量较大,非常适合利用冷冻电镜方法对这个病毒的结构进行重建。”北京大学科学与工程计算中心系统室主任樊春介绍。
据英伟达一份介绍 Tesla P100 GPU 助力冷冻电镜云计算平台的材料,对于一个包含数百万个颗粒,拥有多个构像分子量较大的数据,即使在高性能计算集群上,也可能要花费超过 50 万 CPU 小时的时间。而基于 Tesla P100 GPU 的三维重建,相比于基于 CPU 的计算,有平均大于 15 倍提升。
尽管有高性能计算加速,但由于冷冻电镜依赖大量试验,耗时、耗财、耗人。X 射线晶体学和核磁共振,也有着相似的情况。
因此,不少生物学家转向基于计算机模拟的预测法,采用同源建模或从头预测方式,“猜”出蛋白质三维结构。
从相对准确度来说,最高的是同源建模。顾名思义,为未知结构的蛋白,找到一个与其具有同源性的已知结构蛋白,然后用计算机模拟,根据一级序列预测其三维结构。
对新冠病毒而言,同源性达 80% 左右 、蛋白结构基本已知的 SARS 病毒,自然成为了建模模版。
而在没有已知结构同源蛋白质的情况下,只能采用从头模拟。这种思路不依赖于模版,而是完全根据蛋白质的氨基酸序列来预测。
目前,从头模拟方法最知名的工具,莫过于 DeepMind 旗下的 AlphaFold。作为 AlphaGo 的衍生版本,它曾在 2018 年 12 月第 13 届 CASP 竞赛(以下简称 CASP13 )中获得第一名。这个比赛被誉为蛋白质结构领域的“奥林匹克竞赛”。
在新冠肺炎疫情中,AlphaFold 也利用从头模拟方法,生成了六种可能与新冠病毒有关的蛋白质结构预测结果。相关论文和结果已经发布,但未经过同行评审。

从实现原理上看,AlphaFold 的成功不仅得益于深度学习算法,还有赖于强大的算力支持。
据知乎专业用户郭昊天一个回答,算法上,AlphaFold 是深度学习模型 CNN(卷积神经网络)和传统算法 Rosetta 的结合。实际上,CASP13 前五名都是采用类似的思路。
AlphaFold 之所以能脱颖而出,是因为 TPU 的支持。据中国科学报报道,DeepMind 可以动用几千片 TPU,这是一般科研团队难以比拟的。
TPU 是 Google 为加速神经网络运算能力研发的芯片,处理速度要比 GPU 和 CPU 的组合快 15–30 倍。2014 年 DeepMind 被这家搜索巨头所收购。
DeepMind 没有公布 AlphaFold 用到的 TPU 数量,作为参考,这家公司曾向外界透露,在训练神经网络 BigGAN 时用了 512 块 TPU。BigGAN 以生成的图片质量能以假乱真而闻名。
可见,在预测蛋白质结构领域,算力是决定性因素之一。
除了集中的大规模算力,分散式个人电脑的算力,也被用来支持预测蛋白质结构。3 月中旬,英伟达号召全球玩家来支持分布式计算项目 Folding@home。
这个项目由斯坦福大学于 2000 年发起,是世界上最大的分布式计算项目。每台参与其中的电脑都变成了一个计算节点,当电脑闲置时,算力会被用于模拟蛋白质折叠的过程。
凝聚起来的力量是巨大的。3 月 31 日,Folding@home 项目宣布,已经有超过 100 万的设备加入进来,包括 35.6 万多个英伟达 GPU,7.9 万多个 AMD GPU 以及 59.3 万多个 CPU,算力加起来突破了每秒 100 亿亿。全球最快的超级计算机 Summit,算力峰值也不过每秒 20 亿亿次。

研制药物
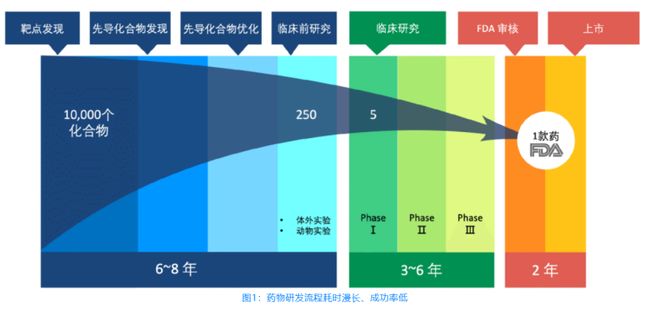
事实上,药物都是被发现的,而不是发明出来的。如上所述,现代制药流程一般是,根据三维结构去筛选适合的化合物。
更具体而言,是先发现苗头化合物,接着据此找到先导化合物,然后转化为候选药物做临床试验。
整个过程十分漫长,据中国科学院一篇名为《高性能计算之源起》的论文,在美国,一种新药上市往往需花费超过 10 亿美元,并耗费 10—17 年的时间。
 (图片来自阿里云)
(图片来自阿里云)
因此,面临一种新疾病,研究人员通常会尝试“老药新用”。“老药”是指已上市或正进行临床试验的药物,“新用”是指发现其新适应症,并将其用于疾病治疗。
在治疗新冠肺炎中,洛匹那韦 / 利托那韦(克力芝)、阿比多尔以及被民众称为“人民的希望”的瑞德西韦等诸多临床用药,都属于老药新用。
传统筛选老药的方法,基本是靠大量生物化学实验,以及临床测试。实验是最保险且不可缺少的环节,但如果完全采取这种方法,时间和人力成本都很高,并且筛选范围有限。
因此,在进行生化实验之前,研究人员会寻求虚拟筛选(Virtual Screening)的支持。
如上文所述,现代制药流程一般是,根据三维结构去筛选适合的药物化合物。虚拟筛选就是通过计算机模拟来寻找化合物,不消耗样品,只需知道蛋白质三维结构即可。
虚拟筛选可分为两类,分别基于受体和基于配体。
前者需要在病毒上找到一个目标靶点(受体),然后根据靶点的蛋白质三维结构,找到活性小分子药物(配体)。后者则是根据已知活性小分子去找到潜在的药物靶点。
这里面有一个药物学的背景知识:大部分药物都是小分子(分子量小于 900 道尔顿)药物。小分子在人体内能较快速地扩散进入细胞,到达作用靶点,通过干扰蛋白间相互作用起效。所谓靶点,即体内具有药效功能,并能被药物作用的部位。这些部位通常是大分子蛋白质。
基于受体进行虚拟筛选,是更加流行的方式,通常会用到两种模拟计算方式:分子对接和分子动力学模拟。
阿里云高性能计算技术专家孙相征介绍,分子对接常用于大量配体的初步筛选,分子动力学模拟用于对分子对接初选结果进一步分析。
据《中华抗生素杂志》一篇论文,分子对接技术是指,通过电脑模拟将小分子放置于大分子靶标的结合区域,再通过计算物理化学参数,预测两者的结合力(结合亲和性)和结合方式(构象),进而找到配体与受体在其活性区域相结合时,能量最低构象的方法。
这个过程,就好像给一把锁找钥匙。蛋白质三维结构就是锁的内部构造,如果完全依靠生化实验,就像亲自将每把候选钥匙都插进锁孔,碰运气看哪把能开。
而借助虚拟筛选技术,可以在计算机模拟开锁过程,让候选钥匙数量减少,最后再把选出来的钥匙亲自做实验。
通常,药物研究公司拥有数量庞大的配体(小分子)库,数量成千上万,甚至更多。
据上述高性能计算专家孙相征计算,如果配体库有 10000 个候选配体,每个配体平均处理时间为 1.5 个小时,总共需要 15000 个小时(625 天)。
分子动力学的模拟计算比分子对接更加耗时。一篇名为《分子对接与分子动力学计算模拟概论》的论文比喻称,如果把分子对接比喻为一幅图片,那么分子动力学模拟就像是一帧桢画面组成的动态电影。
这种方法能将分子动态行为显示到计算机屏幕上,便于直观了解体系在一定条件下的演变过程,广泛应用于材料科学、生物物理和药物设计等领域。
分子拼接和分子动力学模拟背后庞大的计算量,需要超级计算机来支持。据文汇报报道,1 月 3 0 日新冠疫情期间,兰州大学和澳门科技大学的科研工作者,为了进行药物虚拟筛选和分子动力学模拟,联系到上海超级计算中心,希望获得“至少需要 1000 核 CPU,30 块以上英伟达 V100 GPU 加速处理器,以及 50TB 的存储资源”。
目前全球最快的超级计算机系统 Summit,刚刚帮助研究人员从 8000 多种小分子药物化合物中筛选出 77 种。这些化合物能与 S 蛋白结合,阻止病毒表面的分子与 ACE2 受体相连。
Summit 位于美国能源部橡树岭国家实验室,其分子生物物理学中心主任杰瑞米史密斯说:“我们需要通过 Summit 来迅速获得所需的模拟结果,这会花费我们一两天的时间。然而,使用普通计算机则可能花费数月时间。”
不过,正如上文提到,实验依然是不可或缺的环节。通过超级计算机筛选出来的小分子化合物,最后都需要实验来验证其效果和安全性。
总而言之,从揪出病原到摸透蛋白质三维结构,再到研制药物,计算已经是不可或缺的部分。相应地,高性能计算和云计算,成为了让一切加速发生的驱动力。
往期精彩文章推荐



长按关注
品玩官方公众号
科技创新者的每日必读
