复现原文(二):Single-cell RNA sequencing of human


NGS系列文章包括NGS基础、转录组分析 (Nature重磅综述|关于RNA-seq你想知道的全在这)、ChIP-seq分析 (ChIP-seq基本分析流程)、单细胞测序分析 (重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述))、DNA甲基化分析、重测序分析、GEO数据挖掘(典型医学设计实验GEO数据分析 (step-by-step) - Limma差异分析、火山图、功能富集)等内容。
在文章Single-cell RNA sequencing of human kidney中,作者介绍了来自三个人类供体肾脏的23,366个高质量细胞的scRNA-seq数据,并将正常人肾细胞划分为10个clusters。其中,近端肾小管(PT)细胞被分为三个亚型,而集合导管细胞被分为两个亚型。该数据为肾细胞生物学和肾脏疾病的研究提供了可靠的参考。
在本周一的文章(复现原文(一):Single-cell RNA sequencing of human kidney(step by step))中,我们完成了scRNA-seq数据的质控(Hemberg-lab单细胞转录组数据分析(三)- 原始数据质控)、批次校正、找Marker基因(单细胞分群后,怎么找到Marker基因定义每一类群?)、UMAP可视化和tSNE可视化(Hemberg-lab单细胞转录组数据分析(十二)- Scater单细胞表达谱tSNE可视化)。本期我们将提取近端肾小管(PT)细胞来完成下面的后续分析。
#Select a subset of PT cells
PT <- subset(x = kid, idents= c("Proximal convoluted tubule cells","Proximal tubule cells","Proximal straight tubule cells"))
saveRDS(PT,file="PT.rds")
数据准备
加载monocle软件R包:
library(monocle)
monocle输入文件类型有3种类型的格式:
-
表达量文件:
exprs基因在所有细胞中的count值矩阵。 -
表型文件:
phenoData。 -
featureData
3个特征文件转换成CellDataSet格式:
data <- as(as.matrix(PT@assays$RNA@counts), 'sparseMatrix')
pd <- new('AnnotatedDataFrame', data = [email protected])
fData <- data.frame(gene_short_name = row.names(data), row.names = row.names(data))
fd <- new('AnnotatedDataFrame', data = fData)
my_cds <- newCellDataSet(data,
phenoData = pd,
featureData = fd,
lowerDetectionLimit = 0.5,
expressionFamily = negbinomial.size())
对monocle对象进行归一化:
my_cds <- estimateSizeFactors(my_cds)
my_cds <- estimateDispersions(my_cds)
my_cds <- detectGenes(my_cds, min_expr = 0.1) ##过滤掉低质量的基因。
查看数据:
head(fData(my_cds))
![]()
head(pData(my_cds))
![]()
pData(my_cds)$UMI <- Matrix::colSums(exprs(my_cds))
disp_table <- dispersionTable(my_cds)
head(disp_table)
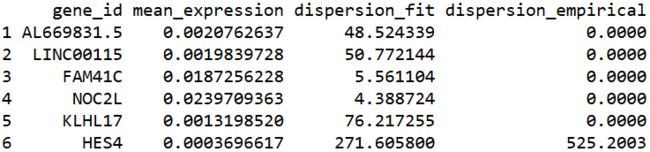
在进行降维聚类之前,先获得高表达的基因集作为每个聚类用来order的Feature基因。也可以使用所有的基因,但一些表达量特别低的基因提供的聚类信号往往会制造分析噪音,Feature基因的选择性很多,一种是可以根据基因的平均表达水平来进行筛选,另外我们也可以选择细胞间异常变异的基因。这些基因往往能较好地反映不同细胞的状态(对一篇单细胞RNA综述的评述:细胞和基因质控参数的选择)。
table(disp_table$mean_expression>=0.1)
unsup_clustering_genes <- subset(disp_table, mean_expression >= 0.1)#细胞平均表达量大于0.1
my_cds <- setOrderingFilter(my_cds, unsup_clustering_genes$gene_id)
plot_ordering_genes(my_cds)

plot_ordering_genes函数显示了基因表达的变异性(分散性)是如何取决于整个细胞的平均表达的。
expressed_genes <- row.names(subset(fData(my_cds), num_cells_expressed >= 10)) #细胞表达个数大于10
my_cds_subset <- my_cds[expressed_genes, ]
my_cds_subset
head(pData(my_cds_subset))
my_cds_subset <- detectGenes(my_cds_subset, min_expr = 0.1)
fData(my_cds_subset)$use_for_ordering <- fData(my_cds_subset)$num_cells_expressed > 0.05 * ncol(my_cds_subset)
table(fData(my_cds_subset)$use_for_ordering)
plot_pc_variance_explained(my_cds_subset, return_all = FALSE)

下面进行降维与聚类
my_cds_subset <- reduceDimension(my_cds_subset,max_components = 2,norm_method = 'log',num_dim = 10,reduction_method = 'tSNE',verbose = TRUE)
my_cds_subset <- clusterCells(my_cds_subset, verbose = FALSE)
plot_rho_delta(my_cds_subset, rho_threshold = 2, delta_threshold = 10)
my_cds_subset <- clusterCells(my_cds_subset,rho_threshold = 2,delta_threshold = 10,skip_rho_sigma = T,verbose = FALSE)
table(pData(my_cds_subset)$Cluster)
plot_cell_clusters(my_cds_subset)
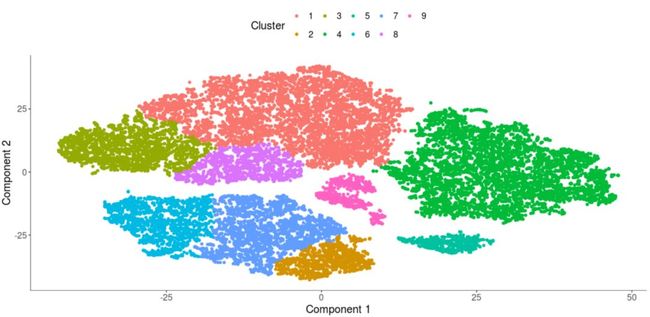
从上图可以看出一种聚成9个clusters。(还在用PCA降维?快学学大牛最爱的t-SNE算法吧, 附Python/R代码)
选择定义细胞发展的基因
head(pData(my_cds_subset))
clustering_DEG_genes <- differentialGeneTest(my_cds_subset,fullModelFormulaStr = '~Cluster',cores = 22)
dim(clustering_DEG_genes)
将细胞按照伪时间排序
library(dplyr)
clustering_DEG_genes %>% arrange(qval) %>% head()
my_ordering_genes <- row.names(clustering_DEG_genes)[order(clustering_DEG_genes$qval)][1:1000]
my_cds_subset <- setOrderingFilter(my_cds_subset, ordering_genes = my_ordering_genes)
my_cds_subset <- reduceDimension(my_cds_subset, method = 'DDRTree') #降维
my_cds_subset <- orderCells(my_cds_subset) #将细胞按照伪时间排序
伪时序轨迹按不同方面绘图
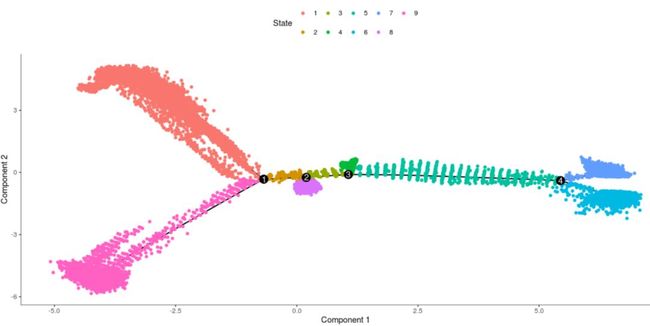
注意参数color_by。(NBT|45种单细胞轨迹推断方法比较,110个实际数据集和229个合成数据集)
plot_cell_trajectory(my_cds_subset, color_by = "State")
plot_cell_trajectory(my_cds_subset, color_by = "RNA_snn_res.0.25")
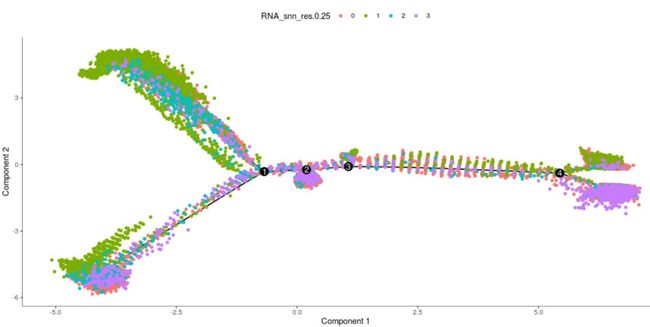
结果图
![]()
原文图
这里由于我们在前期分类的时候使用的是相同的参数,作者分为了3个cluster,我们分成了4个cluster,本质没有什么太大的区别。
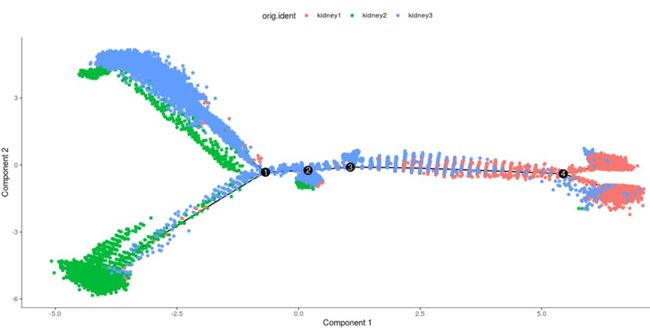
结果图
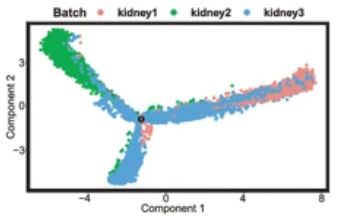
原文图
head(pData(my_cds_subset))
my_pseudotime_de <- differentialGeneTest(my_cds_subset,fullModelFormulaStr = "~sm.ns(Pseudotime)",cores = 22)
my_pseudotime_de %>% arrange(qval) %>% head()
my_pseudotime_de %>% arrange(qval) %>% head() %>% select(gene_short_name) -> my_pseudotime_gene
plot_cell_trajectory(my_cds_subset, color_by = "Pseudotime")

结果图

原文图
影响fate decision的gene变化
#"A" stand for top 6 genes of affecting the fate decisions
A=c("AKR1A1","PDZK1","AKR7A3","AKR7A2","FABP3","GADD45A")
my_pseudotime_gene <-A
plot_genes_in_pseudotime(my_cds_subset[my_pseudotime_gene,])
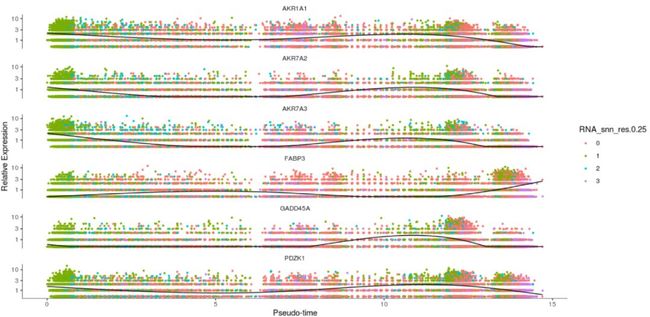
结果图

原文图
从上面我得到的图和原文图的比较来看,结果可能存在一定的差异(…![]()
…),但基本模块是相似的。
整个文章也并没有对以上的伪时序分析进行描述,其最重要的部分应该还是作为人体肾脏单细胞的resource。大家也可以试试呀!
撰文:张虎
校对:生信宝典
参考文献
Liao J, Yu Z, Chen Y, Bao M, Zou C, Zhang H, Liu D, Li T, Zhang Q, Li J, Cheng J, Mo Z. Single-cell RNA sequencing of human kidney. Sci Data. 2020 Jan 2;7(1):4.doi: 10.1038