
enrichplot包实现了几种可视化方法来帮助解释富集结果。它支持从DOSE (Yu et al. 2015)、clusterProfiler (Yu et al. 2012)、ReactomePA (Yu and He 2016)和meshes中获得的可视化富集结果。支持过表达分析(ORA)和基因集富集分析(GSEA)。
enrichplot
包实现了几种可视化方法来帮助解释富集结果。它支持从DOSE (Yu et al. 2015)、clusterProfiler (Yu et al. 2012)、ReactomePA (Yu and He 2016)和meshes中获得的可视化富集结果。支持过表达分析(ORA)和基因集富集分析(GSEA)
Bar Plot
条形图是最广泛使用的可视化丰富项的方法。它将富集分数(如p值)和基因计数或比率描述为条形图的高度和颜色。
library(DOSE)
data(geneList)
de <- names(geneList)[abs(geneList) > 2]
edo <- enrichDGN(de)
library(enrichplot)
barplot(edo, showCategory=20)

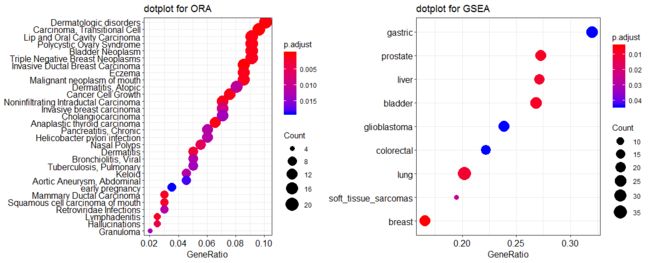
Dot plot
点图类似于条形图,可以将另一个score 作为点的大小。
> edo2 <- gseNCG(geneList, nPerm=10000)
preparing geneSet collections...
GSEA analysis...
leading edge analysis...
done...
Warning messages:
1: In serialize(data, node$con) :
'package:stats' may not be available when loading
2: In serialize(data, node$con) :
'package:stats' may not be available when loading
> p1 <- dotplot(edo, showCategory=30) + ggtitle("dotplot for ORA")
> p2 <- dotplot(edo2, showCategory=30) + ggtitle("dotplot for GSEA")
> plot_grid(p1, p2, ncol=2)
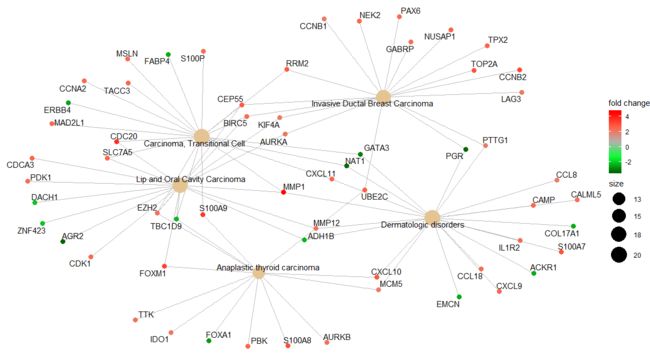
Gene-Concept Network
barplot和dotplot都只显示最显著的富集项,而用户可能想知道哪些基因与这些显著项有关。为了考虑基因可能属于多个注释类别的潜在生物学复杂性,并提供可用的数值变化信息,我们开发了cnetplot函数来提取复杂的关联。cnetplot将基因和生物学概念(例如GO terms或KEGG pathways)之间的联系描述为一个网络。GSEA结果也只支持核心富集基因的表达。
> ## convert gene ID to Symbol
> edox <- setReadable(edo, 'org.Hs.eg.db', 'ENTREZID')
Loading required package: org.Hs.eg.db
Loading required package: AnnotationDbi
Loading required package: stats4
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: ‘BiocGenerics’
The following objects are masked from ‘package:parallel’:
clusterApply, clusterApplyLB, clusterCall,
clusterEvalQ, clusterExport, clusterMap, parApply,
parCapply, parLapply, parLapplyLB, parRapply,
parSapply, parSapplyLB
The following objects are masked from ‘package:stats’:
IQR, mad, sd, var, xtabs
The following objects are masked from ‘package:base’:
anyDuplicated, append, as.data.frame, basename, cbind,
colMeans, colnames, colSums, dirname, do.call,
duplicated, eval, evalq, Filter, Find, get, grep,
grepl, intersect, is.unsorted, lapply, lengths, Map,
mapply, match, mget, order, paste, pmax, pmax.int,
pmin, pmin.int, Position, rank, rbind, Reduce,
rowMeans, rownames, rowSums, sapply, setdiff, sort,
table, tapply, union, unique, unsplit, which,
which.max, which.min
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages
'citation("pkgname")'.
Loading required package: IRanges
Loading required package: S4Vectors
Attaching package: ‘S4Vectors’
The following object is masked from ‘package:base’:
expand.grid
Attaching package: ‘IRanges’
The following object is masked from ‘package:grDevices’:
windows
Warning messages:
1: In is.vector(X) : reached elapsed time limit
2: `keytype` is deprecated. Please use `keyType` instead
> cnetplot(edox, foldChange=geneList)
## categorySize can be scaled by 'pvalue' or 'geneNum'
cnetplot(edox, categorySize="pvalue", foldChange=geneList)

cnetplot(edox, foldChange=geneList, circular = TRUE, colorEdge = TRUE)

如果希望标记节点的子集,可以使用node_label参数,它支持4种可能的选择(即“category”、“gene”、“all”和“none”).
p1 <- cnetplot(edox, node_label="category")
p2 <- cnetplot(edox, node_label="gene")
p3 <- cnetplot(edox, node_label="all")
p4 <- cnetplot(edox, node_label="none")
cowplot::plot_grid(p1, p2, p3, p4, ncol=2, labels=LETTERS[1:4])

Heatmap-like functional classification
热图与cnetplot类似,但将关系显示为热图。如果用户希望显示大量的重要术语,则基因概念网络可能会变得过于复杂。热图可以简化结果,更容易识别表达模式。
heatplot(edox, foldChange=geneList)

Enrichment Map
富集图将被富集的术语组织成一个边缘连接重叠基因集的网络。这样,相互重叠的基因集往往会聚集在一起,使其容易识别功能模块。
emapplot函数支持超几何检验和基因集富集分析的结果。
emapplot(edo)

UpSet Plot
upsetplot是cnetplot的一种替代方法,用于可视化基因和基因集之间的复杂关联。它强调不同基因之间的重叠。
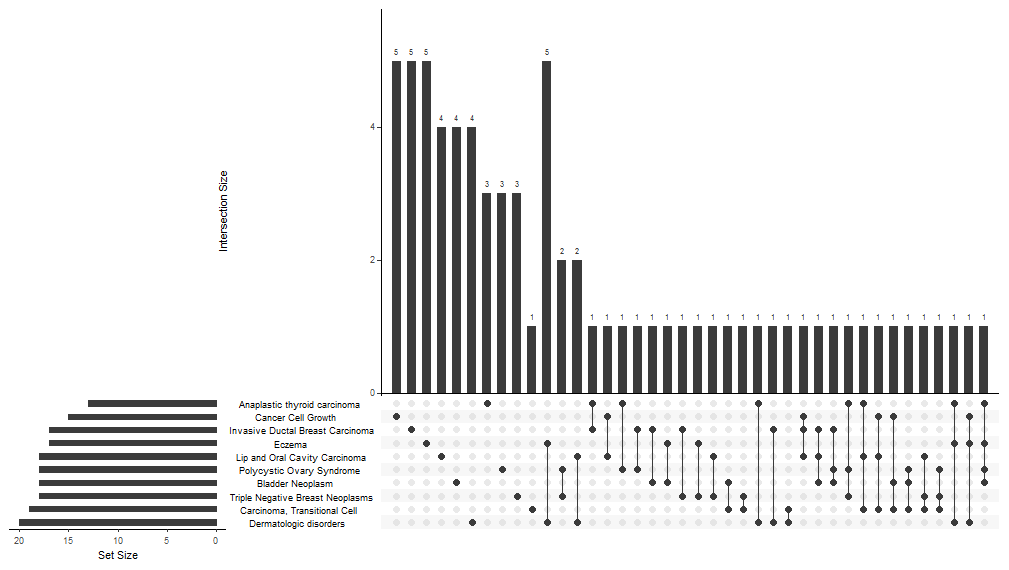
对于过表达分析,upsetplot将计算不同基因集之间的重叠,如图上所示。对于GSEA结果,它将绘制不同类别的褶皱变化分布(例如,路径的独特性,不同路径之间的重叠)。
ridgeline plot for expression distribution of GSEA result
ridgeplot将可视化GSEA富集类别的核心富集基因的表达分布。它帮助用户解释向上/向下调节的路径。
ridgeplot(edo2)

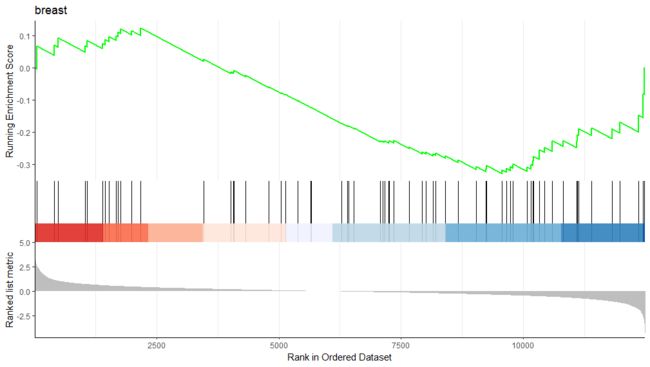
running score and preranked list of GSEA result
运行分数和预先生成的列表是可视化GSEA结果的传统方法。enrichplot
包支持这两者来可视化基因集的分布和富集分数。
gseaplot(edo2, geneSetID = 1, by = "runningScore", title = edo2$Description[1])

gseaplot(edo2, geneSetID = 1, by = "preranked", title = edo2$Description[1])

gseaplot(edo2, geneSetID = 1, title = edo2$Description[1])

gseaplot2(edo2, geneSetID = 1, title = edo2$Description[1])
gseaplot2还支持在同一图中显示多个基因集:
gseaplot2(edo2, geneSetID = 1:3)

gseaplot2(edo2, geneSetID = 1:3, pvalue_table = TRUE,
color = c("#E495A5", "#86B875", "#7DB0DD"), ES_geom = "dot")

library(ggplot2)
library(cowplot)
pp <- lapply(1:3, function(i) {
anno <- edo2[i, c("NES", "pvalue", "p.adjust")]
lab <- paste0(names(anno), "=", round(anno, 3), collapse="\n")
gsearank(edo2, i, edo2[i, 2]) + xlab(NULL) +ylab(NULL) +
annotate("text", 0, edo2[i, "enrichmentScore"] * .9, label = lab, hjust=0, vjust=0)
})
plot_grid(plotlist=pp, ncol=1)

pubmed trend of enriched terms
富集分析的问题之一是寻找进一步研究的途径。在这里,我们提供了pmcplot函数来根据PubMed Central的查询结果来绘制出版物趋势的数量/比例。当然,用户可以在其他场景中使用pmcplot。所有可以在PMC上查询的文本都可以作为pmcplot的输入。
terms <- edo$Description[1:3]
p <- pmcplot(terms, 2010:2017)
p2 <- pmcplot(terms, 2010:2017, proportion=FALSE)
plot_grid(p, p2, ncol=2)

pathview from pathview package
clusterProfiler用户还可以使用pathview中的pathview(Luo和Brouwer 2013)来可视化KEGG路径。
下面的例子说明了如何可视化“hsa04110”通路,这在我们之前的分析中得到了丰富。
library("pathview")
hsa04110 <- pathview(gene.data = geneList,
pathway.id = "hsa04110",
species = "hsa",
limit = list(gene=max(abs(geneList)), cpd=1))

gene-concept-network
疾病和基因注释数据库(DGA, http://dga.nubic.northwestern.edu)是一个合作努力旨在提供一个全面、综合的注释人类基因在疾病网络背景下通过集成可计算的受控词汇表的疾病本体(第三版修订2510,8043继承、发展和获得人类疾病),NCBI基因参考到函数(GeneRIF)和分子相互作用网络(MIN)。DGA使用语义映射将这些资源整合在一起,以建立一套完整的疾病-基因和基因-基因关系,并基于当前的知识提供良好的覆盖率。DGA通过定期重新解析DO、GeneRIF和MINs来保持当前状态。DGA提供了一个用户友好的交互式web界面系统,使用户能够有效地查询、下载和可视化DO树结构和注释,并将其作为树、网络图或表格列表。为了方便集成分析,DGA提供了一个用于与外部分析工具集成的web服务应用程序编程接口。
The Disease and Gene Annotations (DGA): an annotation resource for human disease.