- guava loadingCache代码示例
IM 胡鹏飞
Java工具类介绍
publicclassTest2{publicstaticvoidmain(String[]args)throwsException{LoadingCachecache=CacheBuilder.newBuilder()//设置并发级别为8,并发级别是指可以同时写缓存的线程数.concurrencyLevel(8)//设置缓存容器的初始容量为10.initialCapacity(10)//设置缓存
- 系统学习Python——并发模型和异步编程:进程、线程和GIL
分类目录:《系统学习Python》总目录在文章《并发模型和异步编程:基础知识》我们简单介绍了Python中的进程、线程和协程。本文就着重介绍Python中的进程、线程和GIL的关系。Python解释器的每个实例都是一个进程。使用multiprocessing或concurrent.futures库可以启动额外的Python进程。Python的subprocess库用于启动运行外部程序(不管使用何种
- C++11堆操作深度解析:std::is_heap与std::is_heap_until原理解析与实践
文章目录堆结构基础与函数接口堆的核心性质函数签名与核心接口std::is_heapstd::is_heap_until实现原理深度剖析std::is_heap的验证逻辑std::is_heap_until的定位策略算法优化细节代码实践与案例分析基础用法演示自定义比较器实现最小堆检查边缘情况处理性能分析与实际应用时间复杂度对比典型应用场景与手动实现的对比注意事项与最佳实践迭代器要求比较器设计C++标
- 为什么会出现“与此站点的连接不安全”警告?
当浏览器弹出“与此站点的连接不安全”的红色警告时,不仅会让访客感到不安,还可能直接导致用户流失、品牌信誉受损,甚至引发数据泄露风险。作为网站运营者,如何快速解决这一问题?一、为什么会出现“与此站点的连接不安全”警告?浏览器提示“不安全连接”,本质上是检测到当前网站与用户之间的数据传输未经过加密保护。以下是触发警告的常见原因:1.未安装SSL证书SSL(SecureSocketsLayer)证书是网
- 什么是证书吊销列表?CRL 解释
WoTrusSSL
sslhttps
数字证书是安全在线互动的支柱,用于验证身份和确保加密通信。但是,当这些证书被盗用或滥用时,必须立即撤销它们以维持信任。这就是证书撤销列表(CRL)的作用所在。CRL由证书颁发机构(CA)维护,对于识别和撤销已撤销的证书,防止其造成危害至关重要。在本指南中,我们将探讨什么是CRL、它们如何运作以及为什么它们对网络安全至关重要。什么是证书吊销列表(CRL)?证书吊销列表(CRL)是证书颁发机构(CA)
- 有必要获得WHQL测试认证吗,有什么好处?
什么是WHQL认证?WHQL是MicrosoftWindowsHardwareQualityLab的缩写,中文意思是Windows硬件设备质量实验室,主要是对Windows操作系统的兼容性测试,检验硬件产品和驱动程序在windows系统下的兼容性和稳定性。当某一硬件或软件通过WHQL测试时,制造商可以在其产品包装和广告上使用“DesignedforWindows”标志。该标志可以证明硬件或软件已经
- Flask框架入门:快速搭建轻量级Python网页应用
「已注销」
python-AIpython基础网站网络pythonflask后端
转载:Flask框架入门:快速搭建轻量级Python网页应用1.Flask基础Flask是一个使用Python编写的轻量级Web应用框架。它的设计目标是让Web开发变得快速简单,同时保持应用的灵活性。Flask依赖于两个外部库:Werkzeug和Jinja2,Werkzeug作为WSGI工具包处理Web服务的底层细节,Jinja2作为模板引擎渲染模板。安装Flask非常简单,可以使用pip安装命令
- 驱动程序为什么要做 WHQL 认证?
GDCA SSL证书
网络协议网络
驱动程序进行WHQL(WindowsHardwareQualityLabs)认证的核心价值在于解决兼容性、安全性和市场准入三大关键问题,具体必要性如下:️一、规避系统拦截,保障驱动可用性消除安装警告未认证的驱动在安装时会触发Windows的红色安全警告(如“无法验证发布者”),甚至被系统强制拦截。通过WHQL认证的驱动获得微软数字签名,用户可无阻安装。满足系统强制要求Windows1
- 求是网:“内卷式”竞争的突出表现和主要危害有哪些?
加百力
财经研究科技知识人工智能大数据
"内卷式"竞争主要表现为:企业层面的低价竞争、同质化竞争和营销"逐底竞争";地方政府层面的违规优惠政策、盲目重复建设和设置市场壁垒。危害体现在三个层面:微观上导致"劣币驱逐良币",损害消费者利益;中观上破坏行业生态,挤压产业链利润空间;宏观上扭曲资源配置,抑制创新活力。什么是“内卷式”竞争?概括其一般特征,是指经济主体为了维持市场地位或争夺有限市场,不断投入大量精力和资源,却没有带来整体收益增长的
- WHQL签名怎么申请
GDCA SSL证书
windows
WHQL(WindowsHardwareQualityLabs)签名是微软对硬件和驱动程序进行认证的一种方式,以确保它们与Windows操作系统的兼容性和稳定性。以下是申请WHQL签名的基本步骤,供您参考:1.准备阶段准备硬件设备和驱动程序:确保您的硬件设备已经准备好,并且对应的驱动程序已经经过充分的测试,能够在各种配置和环境下正常工作。获取EV代码签名证书:根据微软的要求,驱动程序进行WHQL认
- JSON 与 AJAX
Auscy
jsonajax前端
一、JSON(JavaScriptObjectNotation)1.数据类型与语法细节支持的数据类型:基本类型:字符串(需用双引号)、数字、布尔值(true/false)、null。复杂类型:数组([])、对象({})。严格语法规范:键名必须用双引号包裹(如"name":"张三")。数组元素用逗号分隔,最后一个元素后不能有多余逗号。数字不能以0开头(如012会被解析为12),不支持八进制/十六进制
- 发票合并工具
小朋的软件园
前端javascriptjavahtml服务器
"发票合并工具"是一款专为高效整理票据设计的实用工具,支持将来自不同渠道的发票文件(如PDF文档、各类图片格式)快速整合为排版规范的PDF文件,尤其适用于财务报销场景下的批量票据处理需求。核心功能亮点多格式兼容:无缝导入PDF文件及常见图片格式(.png/.jpg/.jpeg/.bmp),适配多来源发票整合需求。智能布局配置:提供灵活的页面布局选项(每页2/3/4张发票),其中"2合1"模式针对报
- Python Flask 框架入门:快速搭建 Web 应用的秘诀
Python编程之道
Python人工智能与大数据Python编程之道pythonflask前端ai
PythonFlask框架入门:快速搭建Web应用的秘诀关键词Flask、微框架、路由系统、Jinja2模板、请求处理、WSGI、Web开发摘要想快速用Python搭建一个灵活的Web应用?Flask作为“微框架”代表,凭借轻量、可扩展的特性,成为初学者和小型项目的首选。本文将从Flask的核心概念出发,结合生活化比喻、代码示例和实战案例,带你一步步掌握:如何用Flask搭建第一个Web应用?路由
- C++ 11 Lambda表达式和min_element()与max_element()的使用_c++ lamda函数 min_element((1)
2401_84976182
程序员c语言c++学习
既有适合小白学习的零基础资料,也有适合3年以上经验的小伙伴深入学习提升的进阶课程,涵盖了95%以上CC++开发知识点,真正体系化!由于文件比较多,这里只是将部分目录截图出来,全套包含大厂面经、学习笔记、源码讲义、实战项目、大纲路线、讲解视频,并且后续会持续更新如果你需要这些资料,可以戳这里获取#include#include#includeusingnamespacestd;boolcmp(int
- C++ 11 Lambda表达式和min_element()与max_element()的使用_c++ lamda函数 min_element(
网上学习资料一大堆,但如果学到的知识不成体系,遇到问题时只是浅尝辄止,不再深入研究,那么很难做到真正的技术提升。需要这份系统化的资料的朋友,可以添加戳这里获取一个人可以走的很快,但一群人才能走的更远!不论你是正从事IT行业的老鸟或是对IT行业感兴趣的新人,都欢迎加入我们的的圈子(技术交流、学习资源、职场吐槽、大厂内推、面试辅导),让我们一起学习成长!intmain(){vectormyvec{3,
- k8s:安装 Helm 私有仓库ChartMuseum、helm-push插件并上传、安装Zookeeper
云游
dockerhelmhelm-push
ChartMuseum是Kubernetes生态中用于存储、管理和发布HelmCharts的开源系统,主要用于扩展Helm包管理器的功能核心功能集中存储:提供中央化仓库存储Charts,支持版本管理和权限控制。跨集群部署:支持多集群环境下共享Charts,简化部署流程。离线部署:适配无网络环境,可将Charts存储在本地或局域网内。HTTP接口:通过HTTP协议提供服务,用户
- 上位机知识篇---SD卡&U盘镜像
常用的镜像烧录软件balenaEtcherbalenaEtcher是一个开源的、跨平台的工具,用于将操作系统镜像文件(如ISO和IMG文件)烧录到SD卡和USB驱动器中。以下是其使用方法、使用场景和使用注意事项的介绍:使用方法下载安装:根据自己的操作系统,从官方网站下载对应的安装包。Windows系统下载.exe文件后双击安装;Linux系统若下载的是.deb文件,可在终端执行“sudodpkg-
- 【LeetCode 热题 100】24. 两两交换链表中的节点——(解法一)迭代+哨兵
xumistore
LeetCodeleetcode链表算法java
Problem:24.两两交换链表中的节点题目:给你一个链表,两两交换其中相邻的节点,并返回交换后链表的头节点。你必须在不修改节点内部的值的情况下完成本题(即,只能进行节点交换)。文章目录整体思路完整代码时空复杂度时间复杂度:O(N)空间复杂度:O(1)整体思路这段代码旨在解决一个经典的链表操作问题:两两交换链表中的节点(SwapNodesinPairs)。问题要求将链表中每两个相邻的节点进行交换
- Guava LoadingCache
sqyaa.
java并发编程Java知识jvm缓存guava
LoadingCache是GoogleGuava库提供的一个高级缓存实现,它通过自动加载机制简化了缓存使用模式。核心特性自动加载机制当缓存未命中时,自动调用指定的CacheLoader加载数据线程安全:并发请求下,相同key只会加载一次灵活的过期策略支持基于写入时间(expireAfterWrite)和访问时间(expireAfterAccess)的过期可设置最大缓存大小,基于LRU策略淘汰丰富的
- JavaScript 树形菜单总结
Auscy
microsoft
树形菜单是前端开发中常见的交互组件,用于展示具有层级关系的数据(如文件目录、分类列表、组织架构等)。以下从核心概念、实现方式、常见功能及优化方向等方面进行总结。一、核心概念层级结构:数据以父子嵌套形式存在,如{id:1,children:[{id:2}]}。节点:树形结构的基本单元,包含自身信息及子节点(若有)。展开/折叠:子节点的显示与隐藏切换,是树形菜单的核心交互。递归渲染:因数据层级不固定,
- 基于定制开发开源AI智能名片S2B2C商城小程序的社群游戏定制策略研究
说私域
人工智能小程序游戏
摘要:本文聚焦社群游戏定制领域,深入探讨以社群文化和用户偏好为导向的定制策略。通过分析互动游戏活动、社群文化塑造等关键要素,结合定制开发开源AI智能名片S2B2C商城小程序的技术特性,提出针对性游戏定制方案。研究旨在提升社群用户参与度与游戏体验,为社群游戏发展提供理论支持与实践指导。关键词:社群游戏定制;定制开发开源AI智能名片S2B2C商城小程序;社群文化;用户偏好一、引言在数字化社交蓬勃发展的
- 冒泡、选择、插入排序:三大基础排序算法深度解析(C语言实现)
xienda
算法排序算法数据结构
在算法学习道路上,排序算法是每位程序员必须掌握的基石。本文将深入解析冒泡排序、选择排序和插入排序这三种基础排序算法,通过C语言代码实现和对比分析,帮助读者彻底理解它们的差异与应用场景。算法原理与代码实现1.冒泡排序(BubbleSort)工作原理:通过重复比较相邻元素,将较大元素逐步"冒泡"到数组末尾。voidbubbleSort(intarr[],intn){ for(inti=0;iarr[
- Leetcode 148. 排序链表
文章目录前引题目代码(首刷看题解)代码(8.9二刷部分看解析)代码(9.15三刷部分看解析)前引综合性比较强的一道题,要求时间复杂度必须O(logn)才能通过,最适合链表的排序算法就是归并。这里采用自顶向下的方法步骤:找到链表中点(双指针)对两个子链表排序(递归,直到只有一个结点,记得将子链表最后指向nullptr)归并(引入dummy结点)题目Leetcode148.排序链表代码(首刷看题解)c
- 全面触摸屏输入法设计与实现
长野君
本文还有配套的精品资源,点击获取简介:触摸屏输入法是针对触摸设备优化的文字输入方案,包括虚拟键盘、手写、语音识别和手势等多种输入方式。本方案通过提供主程序文件、用户手册、界面截图、示例图、说明文本和音效文件,旨在为用户提供一个完整的、多样的文字输入体验。开发者通过持续优化算法和用户界面,使用户在无物理键盘环境下也能高效准确地进行文字输入。1.触摸屏输入法概述简介在现代信息技术飞速发展的今天,触摸屏
- LeetCode 148. 排序链表:归并排序的细节解析
进击的小白菜
2025Top100详解leetcode链表算法
文章目录题目描述一、方法思路:归并排序的核心步骤二、关键实现细节:快慢指针分割链表1.快慢指针的初始化问题2.为什么选择`fast=head.next`?示例1:链表长度为偶数(`1->2->3->4`)三、完整代码实现四、复杂度分析五、总结题目描述LeetCode148题要求对链表进行排序,时间复杂度需为O(nlogn),且空间复杂度为O(logn)。由于链表的特殊结构(无法随机访问),归并排序
- 前端项目架构设计要领
1.架构设计的核心目标在设计前端项目架构时,核心目标是模块化、可维护、可扩展、可测试,以及开发效率的最大化。这些目标可以通过以下几个方面来实现:组件化:将UI功能封装为可复用的组件。模块化:将业务逻辑分解为独立的模块或服务。自动化构建与部署:实现自动化构建、测试和部署流程,减少人为操作的错误。代码规范化与检查:确保团队协作时,代码风格和质量一致。2.项目目录结构设计一个清晰合理的目录结构对大型项目
- 精通Canvas:15款时钟特效代码实现指南
烟幕缭绕
本文还有配套的精品资源,点击获取简介:HTML5的Canvas是一个用于绘制矢量图形的API,通过JavaScript实现动态效果。本项目集合了15种不同的时钟特效代码,帮助开发者通过学习绘制圆形、线条、时间更新、旋转、颜色样式设置及动画效果等概念,深化对Canvas的理解和应用。项目中的CSS文件负责时钟的样式设定,而JS文件则包含实现各种特效的逻辑,通过不同的函数或类处理时间更新和动画绘制,提
- 高效批量单词翻译工具的设计与应用
本文还有配套的精品资源,点击获取简介:在信息技术飞速发展的今天,批量单词翻译工具通过计算机的数据处理能力,大大提高了语言学习和文字处理的效率。用户通过简单输入单词列表到一个文本文件,并运行翻译程序,即可获得翻译结果并保存至指定文件。该工具集成了内置或外部翻译引擎,利用自然语言处理技术实现快速准确的翻译,并可能提供词性识别等附加功能。尽管机器翻译无法完全取代人工校对,但它为用户提供了一种高效的翻译解
- 嵌入式系统LCD显示模块编程实践
本文还有配套的精品资源,点击获取简介:本文档提供了一个具有800x480分辨率的3.5英寸液晶显示模块LW350AC9001的驱动程序代码,以及嵌入式系统中使用C/C++语言进行硬件编程的实践指南。该模块的2mm厚度使其适用于空间受限的便携式设备。内容包括驱动程序源代码、硬件控制接口使用方法,以及如何在嵌入式系统中进行图形处理、电源管理与性能优化。1.嵌入式系统原理1.1嵌入式系统概念嵌入式系统是
- 深入剖析OpenJDK 18 GA源码:Java平台最新发展
想法臃肿
本文还有配套的精品资源,点击获取简介:OpenJDK18GA作为Java开发的关键里程碑,提供了诸多新特性和改进。本文章深入探讨了OpenJDK18GA源码,揭示其内部机制,帮助开发者更好地理解和利用这个版本。文章还涵盖了PatternMatching、SealedClasses、Records、JEP395、JEP406和JEP407等特性,以及HotSpot虚拟机、编译器、垃圾收集器、内存模型
- 开发者关心的那些事
圣子足道
ios游戏编程apple支付
我要在app里添加IAP,必须要注册自己的产品标识符(product identifiers)。产品标识符是什么?
产品标识符(Product Identifiers)是一串字符串,它用来识别你在应用内贩卖的每件商品。App Store用产品标识符来检索产品信息,标识符只能包含大小写字母(A-Z)、数字(0-9)、下划线(-)、以及圆点(.)。你可以任意排列这些元素,但我们建议你创建标识符时使用
- 负载均衡器技术Nginx和F5的优缺点对比
bijian1013
nginxF5
对于数据流量过大的网络中,往往单一设备无法承担,需要多台设备进行数据分流,而负载均衡器就是用来将数据分流到多台设备的一个转发器。
目前有许多不同的负载均衡技术用以满足不同的应用需求,如软/硬件负载均衡、本地/全局负载均衡、更高
- LeetCode[Math] - #9 Palindrome Number
Cwind
javaAlgorithm题解LeetCodeMath
原题链接:#9 Palindrome Number
要求:
判断一个整数是否是回文数,不要使用额外的存储空间
难度:简单
分析:
题目限制不允许使用额外的存储空间应指不允许使用O(n)的内存空间,O(1)的内存用于存储中间结果是可以接受的。于是考虑将该整型数反转,然后与原数字进行比较。
注:没有看到有关负数是否可以是回文数的明确结论,例如
- 画图板的基本实现
15700786134
画图板
要实现画图板的基本功能,除了在qq登陆界面中用到的组件和方法外,还需要添加鼠标监听器,和接口实现。
首先,需要显示一个JFrame界面:
public class DrameFrame extends JFrame { //显示
- linux的ps命令
被触发
linux
Linux中的ps命令是Process Status的缩写。ps命令用来列出系统中当前运行的那些进程。ps命令列出的是当前那些进程的快照,就是执行ps命令的那个时刻的那些进程,如果想要动态的显示进程信息,就可以使用top命令。
要对进程进行监测和控制,首先必须要了解当前进程的情况,也就是需要查看当前进程,而 ps 命令就是最基本同时也是非常强大的进程查看命令。使用该命令可以确定有哪些进程正在运行
- Android 音乐播放器 下一曲 连续跳几首歌
肆无忌惮_
android
最近在写安卓音乐播放器的时候遇到个问题。在MediaPlayer播放结束时会回调
player.setOnCompletionListener(new OnCompletionListener() {
@Override
public void onCompletion(MediaPlayer mp) {
mp.reset();
Log.i("H
- java导出txt文件的例子
知了ing
javaservlet
代码很简单就一个servlet,如下:
package com.eastcom.servlet;
import java.io.BufferedOutputStream;
import java.io.IOException;
import java.net.URLEncoder;
import java.sql.Connection;
import java.sql.Resu
- Scala stack试玩, 提高第三方依赖下载速度
矮蛋蛋
scalasbt
原文地址:
http://segmentfault.com/a/1190000002894524
sbt下载速度实在是惨不忍睹, 需要做些配置优化
下载typesafe离线包, 保存为ivy本地库
wget http://downloads.typesafe.com/typesafe-activator/1.3.4/typesafe-activator-1.3.4.zip
解压r
- phantomjs安装(linux,附带环境变量设置) ,以及casperjs安装。
alleni123
linuxspider
1. 首先从官网
http://phantomjs.org/下载phantomjs压缩包,解压缩到/root/phantomjs文件夹。
2. 安装依赖
sudo yum install fontconfig freetype libfreetype.so.6 libfontconfig.so.1 libstdc++.so.6
3. 配置环境变量
vi /etc/profil
- JAVA IO FileInputStream和FileOutputStream,字节流的打包输出
百合不是茶
java核心思想JAVA IO操作字节流
在程序设计语言中,数据的保存是基本,如果某程序语言不能保存数据那么该语言是不可能存在的,JAVA是当今最流行的面向对象设计语言之一,在保存数据中也有自己独特的一面,字节流和字符流
1,字节流是由字节构成的,字符流是由字符构成的 字节流和字符流都是继承的InputStream和OutPutStream ,java中两种最基本的就是字节流和字符流
类 FileInputStream
- Spring基础实例(依赖注入和控制反转)
bijian1013
spring
前提条件:在http://www.springsource.org/download网站上下载Spring框架,并将spring.jar、log4j-1.2.15.jar、commons-logging.jar加载至工程1.武器接口
package com.bijian.spring.base3;
public interface Weapon {
void kil
- HR看重的十大技能
bijian1013
提升能力HR成长
一个人掌握何种技能取决于他的兴趣、能力和聪明程度,也取决于他所能支配的资源以及制定的事业目标,拥有过硬技能的人有更多的工作机会。但是,由于经济发展前景不确定,掌握对你的事业有所帮助的技能显得尤为重要。以下是最受雇主欢迎的十种技能。 一、解决问题的能力 每天,我们都要在生活和工作中解决一些综合性的问题。那些能够发现问题、解决问题并迅速作出有效决
- 【Thrift一】Thrift编译安装
bit1129
thrift
什么是Thrift
The Apache Thrift software framework, for scalable cross-language services development, combines a software stack with a code generation engine to build services that work efficiently and s
- 【Avro三】Hadoop MapReduce读写Avro文件
bit1129
mapreduce
Avro是Doug Cutting(此人绝对是神一般的存在)牵头开发的。 开发之初就是围绕着完善Hadoop生态系统的数据处理而开展的(使用Avro作为Hadoop MapReduce需要处理数据序列化和反序列化的场景),因此Hadoop MapReduce集成Avro也就是自然而然的事情。
这个例子是一个简单的Hadoop MapReduce读取Avro格式的源文件进行计数统计,然后将计算结果
- nginx定制500,502,503,504页面
ronin47
nginx 错误显示
server {
listen 80;
error_page 500/500.html;
error_page 502/502.html;
error_page 503/503.html;
error_page 504/504.html;
location /test {return502;}}
配置很简单,和配
- java-1.二叉查找树转为双向链表
bylijinnan
二叉查找树
import java.util.ArrayList;
import java.util.List;
public class BSTreeToLinkedList {
/*
把二元查找树转变成排序的双向链表
题目:
输入一棵二元查找树,将该二元查找树转换成一个排序的双向链表。
要求不能创建任何新的结点,只调整指针的指向。
10
/ \
6 14
/ \
- Netty源码学习-HTTP-tunnel
bylijinnan
javanetty
Netty关于HTTP tunnel的说明:
http://docs.jboss.org/netty/3.2/api/org/jboss/netty/channel/socket/http/package-summary.html#package_description
这个说明有点太简略了
一个完整的例子在这里:
https://github.com/bylijinnan
- JSONUtil.serialize(map)和JSON.toJSONString(map)的区别
coder_xpf
jqueryjsonmapval()
JSONUtil.serialize(map)和JSON.toJSONString(map)的区别
数据库查询出来的map有一个字段为空
通过System.out.println()输出 JSONUtil.serialize(map): {"one":"1","two":"nul
- Hibernate缓存总结
cuishikuan
开源sshjavawebhibernate缓存三大框架
一、为什么要用Hibernate缓存?
Hibernate是一个持久层框架,经常访问物理数据库。
为了降低应用程序对物理数据源访问的频次,从而提高应用程序的运行性能。
缓存内的数据是对物理数据源中的数据的复制,应用程序在运行时从缓存读写数据,在特定的时刻或事件会同步缓存和物理数据源的数据。
二、Hibernate缓存原理是怎样的?
Hibernate缓存包括两大类:Hib
- CentOs6
dalan_123
centos
首先su - 切换到root下面1、首先要先安装GCC GCC-C++ Openssl等以来模块:yum -y install make gcc gcc-c++ kernel-devel m4 ncurses-devel openssl-devel2、再安装ncurses模块yum -y install ncurses-develyum install ncurses-devel3、下载Erang
- 10款用 jquery 实现滚动条至页面底端自动加载数据效果
dcj3sjt126com
JavaScript
无限滚动自动翻页可以说是web2.0时代的一项堪称伟大的技术,它让我们在浏览页面的时候只需要把滚动条拉到网页底部就能自动显示下一页的结果,改变了一直以来只能通过点击下一页来翻页这种常规做法。
无限滚动自动翻页技术的鼻祖是微博的先驱:推特(twitter),后来必应图片搜索、谷歌图片搜索、google reader、箱包批发网等纷纷抄袭了这一项技术,于是靠滚动浏览器滚动条
- ImageButton去边框&Button或者ImageButton的背景透明
dcj3sjt126com
imagebutton
在ImageButton中载入图片后,很多人会觉得有图片周围的白边会影响到美观,其实解决这个问题有两种方法
一种方法是将ImageButton的背景改为所需要的图片。如:android:background="@drawable/XXX"
第二种方法就是将ImageButton背景改为透明,这个方法更常用
在XML里;
<ImageBut
- JSP之c:foreach
eksliang
jspforearch
原文出自:http://www.cnblogs.com/draem0507/archive/2012/09/24/2699745.html
<c:forEach>标签用于通用数据循环,它有以下属性 属 性 描 述 是否必须 缺省值 items 进行循环的项目 否 无 begin 开始条件 否 0 end 结束条件 否 集合中的最后一个项目 step 步长 否 1
- Android实现主动连接蓝牙耳机
gqdy365
android
在Android程序中可以实现自动扫描蓝牙、配对蓝牙、建立数据通道。蓝牙分不同类型,这篇文字只讨论如何与蓝牙耳机连接。
大致可以分三步:
一、扫描蓝牙设备:
1、注册并监听广播:
BluetoothAdapter.ACTION_DISCOVERY_STARTED
BluetoothDevice.ACTION_FOUND
BluetoothAdapter.ACTION_DIS
- android学习轨迹之四:org.json.JSONException: No value for
hyz301
json
org.json.JSONException: No value for items
在JSON解析中会遇到一种错误,很常见的错误
06-21 12:19:08.714 2098-2127/com.jikexueyuan.secret I/System.out﹕ Result:{"status":1,"page":1,&
- 干货分享:从零开始学编程 系列汇总
justjavac
编程
程序员总爱重新发明轮子,于是做了要给轮子汇总。
从零开始写个编译器吧系列 (知乎专栏)
从零开始写一个简单的操作系统 (伯乐在线)
从零开始写JavaScript框架 (图灵社区)
从零开始写jQuery框架 (蓝色理想 )
从零开始nodejs系列文章 (粉丝日志)
从零开始编写网络游戏
- jquery-autocomplete 使用手册
macroli
jqueryAjax脚本
jquery-autocomplete学习
一、用前必备
官方网站:http://bassistance.de/jquery-plugins/jquery-plugin-autocomplete/
当前版本:1.1
需要JQuery版本:1.2.6
二、使用
<script src="./jquery-1.3.2.js" type="text/ja
- PLSQL-Developer或者Navicat等工具连接远程oracle数据库的详细配置以及数据库编码的修改
超声波
oracleplsql
在服务器上将Oracle安装好之后接下来要做的就是通过本地机器来远程连接服务器端的oracle数据库,常用的客户端连接工具就是PLSQL-Developer或者Navicat这些工具了。刚开始也是各种报错,什么TNS:no listener;TNS:lost connection;TNS:target hosts...花了一天的时间终于让PLSQL-Developer和Navicat等这些客户
- 数据仓库数据模型之:极限存储--历史拉链表
superlxw1234
极限存储数据仓库数据模型拉链历史表
在数据仓库的数据模型设计过程中,经常会遇到这样的需求:
1. 数据量比较大; 2. 表中的部分字段会被update,如用户的地址,产品的描述信息,订单的状态等等; 3. 需要查看某一个时间点或者时间段的历史快照信息,比如,查看某一个订单在历史某一个时间点的状态, 比如,查看某一个用户在过去某一段时间内,更新过几次等等; 4. 变化的比例和频率不是很大,比如,总共有10
- 10点睛Spring MVC4.1-全局异常处理
wiselyman
spring mvc
10.1 全局异常处理
使用@ControllerAdvice注解来实现全局异常处理;
使用@ControllerAdvice的属性缩小处理范围
10.2 演示
演示控制器
package com.wisely.web;
import org.springframework.stereotype.Controller;
import org.spring

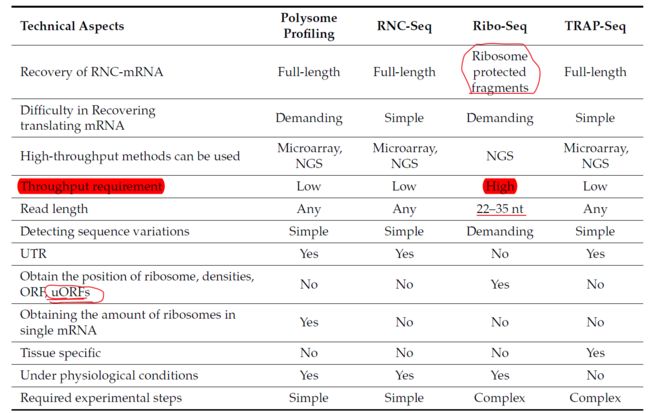
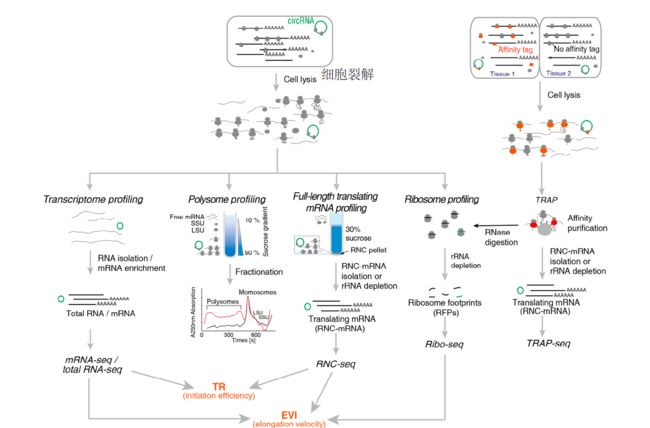 The major translatomic methods which investigate translating RNA
The major translatomic methods which investigate translating RNA