网址点这里,根据网上资料进行的学习,可能有些操作有变动。
设置工作目录,载入必要的包
source("https://bioconductor.org/biocLite.R")
biocLite()
getwd()
setwd("D:/RWork/Rlearning")
library(GEOquery)
biocLite("Rcpp")
然后就有报错:Warning message:package ‘Rcpp’ is not available (for R version 3.4.2)。有回答说可能是镜像源的问题,我想起来前天把自动的源换成国内中科大的源了,只得再改回去(第一个,`Global(CDN)-RStudio)。

重新运行一遍,可以了,继续。
下载数据,并且解压
x = getGEOSuppFiles("GSE20986")
下载的是原文件的压缩文件。有相同功能的函数还有getGEO。
解压文件,解压到“./data”里面去了,然后再解压文件,得到CEL文件。
untar("GSE20986/GSE20986_RAW.tar", exdir = "data")
cels = list.files("data/", pattern = "[gz]")
sapply(paste("data", cels, sep = "/"), gunzip)
创建phenodata
创建phenodata文件,返回的结果是[1] "matrix"。
phenodata = matrix(rep(list.files("data", pattern= "CEL"), 2), ncol =2)
class(phenodata)
phenodata储存的是样本的分组信息。
phenodata <- as.data.frame(phenodata) #phenodata的类转换成了data.frame
colnames(phenodata) <- c("Name", "FileName") #为phenodata命名了列名
关于R的数据结构,将会另开一篇详述。此处数据框增加了一列"Target"储存样本分组信息,可以在GEO数据库中找到分组信息。最后phenodata写入了/GSE20986phenodata.txt文件中。
phenodata$Targets <- c("iris", "retina", "retina", "iris", "retina", "iris",
"choroid", "choroid", "choroid", "huvec", "huvec",
"huvec")
write.table(phenodata, "data/GSE20986phenodata.txt", quote = F, sep = "\t", row.names = F)
原始数据初步处理
library(simleaffy)
celfiles <- read.affy(covdesc = "GSE20986phenodata.txt", path = "data")
#注意这一步phenodata必须放在"data"路径下面,我也不知道为什么。。。。
#另外在phenodata数据框创建过程中,应该对rownames进行命名,可用>rownames(phenodata <- phenodata$Name)
boxplot(celfiles)
上面的语句用simpleaffy里的read.affy函数生成了一个AffyBatch对象。函数read.affy的第一个参数covdesc需要一个描述样本分组的数据框文件,第一列是CEL文件(路径)名,其他列是实验中各的因子变量值,这里是phenodata$Targets。生成图像如下;

标定颜色
library(RColorBrewer)
cols = brewer.pal(8, "Set1")
eset <- exprs(celfiles)
samples <- celfiles$Targets
colnames(eset)
特异查看了一下col的内容,返回如下[1] "#E41A1C" "#377EB8" "#4DAF4A" "#984EA3" "#FF7F00" "#FFFF33" "#A65628" "#F781BF",是一系列(8个)颜色值构成的字符串向量。上面的语句将AffyBatch类的实例celfiles用函数exprs转换成了matrix类的实例eset。矩阵的列名为样本名:[1] "GSM524662.CEL" "GSM524663.CEL" "GSM524664.CEL" "GSM524665.CEL" "GSM524666.CEL" [6] "GSM524667.CEL" "GSM524668.CEL" "GSM524669.CEL" "GSM524670.CEL" "GSM524671.CEL" [11] "GSM524672.CEL" "GSM524673.CEL"。接着用字符串向量samples装载了样本的分组信息。
对celfies传递颜色参数cols后,图像漂亮多了。
boxplot(celfiles, col = cols, las = 2)

将分组的信息传递给 eset
colnames(eset) <- samples
看一下此时的eset

预处理
第一步加载必要的包:
require(simpleaffy)
require(affyPLM)
#载入这个函数是为了对标准化生成的对象celfiles.gcrma数据进行解析
下面进行的是预处理,使用的函数是gcrma,接受对象是AffyBatch类实例celfiles,返回的是ExpressionSet类对象,对象名celfiles.gcrma。
celfiles.gcrma = gcrma(celfiles)
gcrma函数采用的是GCRMA算法,RMA算法原理介绍。gcrma算法暂时未查到资料,待解决。
对处理后的数据进行绘图并与处理前对比。
par(mfrow=c(1,2))
boxplot(celfiles.gcrma, col = cols, las = 2, main = "Post-Normalization");
boxplot(celfiles, col = cols, las = 2, main = "Pre-Normalization")
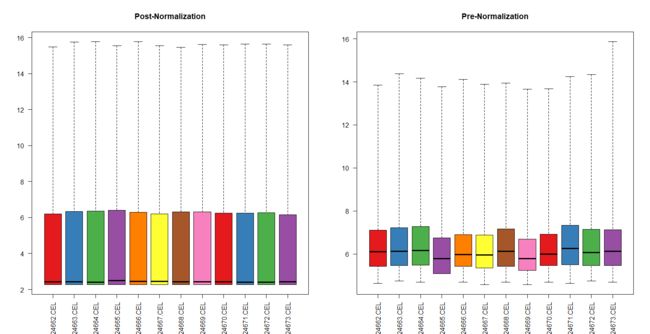
然后我们对样本再进行聚类:
dev.off() #清除图像输出参数
distance <- dist(t(exprs(celfiles.gcrma)), method = "maximum")
clusters <- hclust(distance)
plot(clusters)
结果如下:

这里我们看一下另外一个实验的标准化处理前后的样本表达强度的变化:
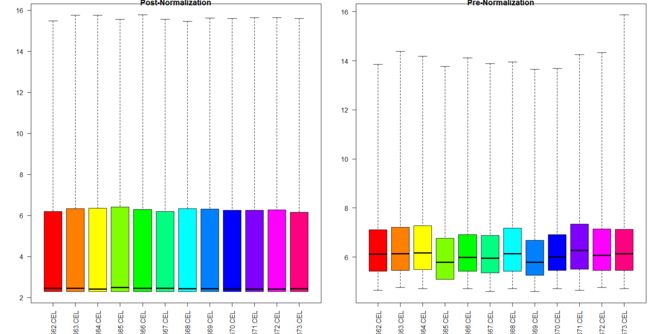
聚类树
先上代码:
distance <- dist(t(eset), method = "maximum")
clusters <- hclust(distance)
plot(clusters)
第一次接触该函数,根据帮助文档dist是一个接受矩阵对象按一定距离计算方法计算行之间的距离并返回距离矩阵的函数,距离计算方法是 maximum ,是两元素 x 和 y 的最大距离。函数返回的是一个dist,距离矩阵,内容如下:

hclust是一个进行分层聚类分析的函数,接受 hist实例,返回 hclust实例,描述了聚类所生成的 树。结果描述如下:
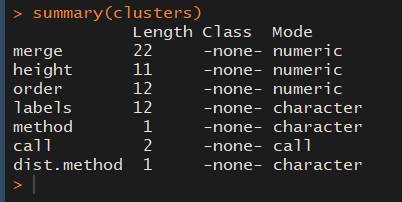
画图所得结果如下:

计算表达值
library(limma)
查看我们的phenodata对象:
phenodata

下面的语句将 samples变成一个因子变量,并创建方案设计矩阵 design:
samples <- as.factor(samples)
design <- model.matrix(~0+samples)
查看前后变化如图:
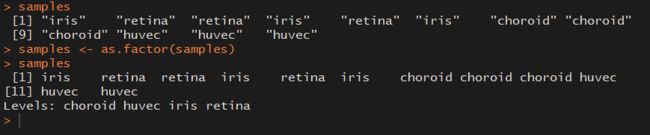
对 design的列名进行重命名:
colnames(design) <- c("choroid", "huvec", "iris", "retina")
design
下面的语句进行了对比设计,然后用贝叶斯检验计算了探针矩阵的差异表达,最后一句加载了一个注释包。
contrast.matrix = makeContrasts(
huvec_choroid = huvec - choroid,
huvec_retina = huvec - retina,
huvec_iris <- huvec - iris,
levels = design)
fit = lmFit(celfiles.gcrma, design) #返回 [1] "MArrayLM"
huvec_fit <- contrasts.fit(fit, contrast.matrix)
huvec_ebay <- eBayes(huvec_fit)
library(hgu133plus2.db)
下面的语句进行了geneSymbol的映射。
probenames.list <- rownames(topTable(huvec_ebay, number = 100000))
getsymbols <- getSYMBOL(probenames.list, "hgu133plus2")
results <- topTable(huvec_ebay, number = 100000, coef = "huvec_choroid")
results <- cbind(results, getsymbols)

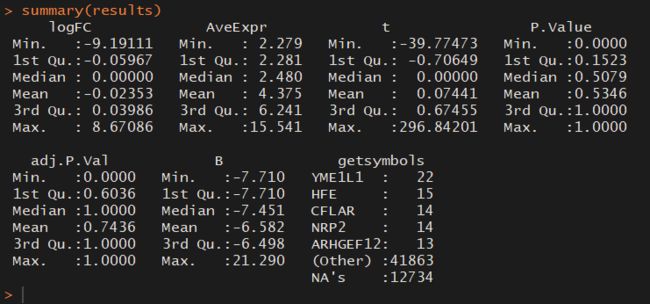
取定阈值
results$threshold <- "1"
a <- subset(results, adj.P.Val < 0.05 & logFC > 5)
results[rownames(a), "threshold"] <- "2"
b <- subset(results, adj.P.Val < 0.05 & logFC < -5)
results[rownames(b), "threshold"] <- "3"
table(results$threshold)
绘图
library(ggplot2)
volcano <- ggplot(data = results,
aes(x = logFC, y = -1*log10(adj.P.Val),
colour = threshold,
label = getsymbols))
volcano <- volcano +
geom_point() +
scale_color_manual(values = c("black", "red", "green"),
labels = c("Not Significant", "Upregulated", "Downregulated"),
name = "Key/Legend")
volcano +
geom_text(data = subset(results, logFC > 5 & -1*log10(adj.P.Val) > 5), aes(x = logFC, y = -1*log10(adj.P.Val), colour = threshold, label = getsymbols) )
结果如图:

在这一步里面我们可以看到筛选的条件是
adj.P.Val < 0.05 & logFC > 5,没有使用常规的
LogFC > 2也没有去除没有基因名
results$getsymbols == NA的探针行,下面的语句就是进行了这些筛选,并把结果保存在了
GSE20986.txt文档中,后期可用excel表格提取出符合条件的基因进行画图和富集分析。
resultsSave <- subset(results, adj.P.Val < 0.05 & abs(logFC) > 1 & !is.na(getsymbols))
write.csv(resultsSave[,], file = "GSE20986.txt")
得到结果如图:

分别获取上调和下调基因
up_genes <- subset(resulstSave, resultsSave$logFC > 0)
down_genes <- subset(resultsSave, resultsSave$logFC < 0)
write.table(up_genes, file = "GSE20986upGenes.txt", sep = "\t")
write.table(down_genes, file = "GSE20986downGenes.txt", sep = "\t")
后面就是分别对这些up和down的基因分别进行GO和KEGG分析啦!