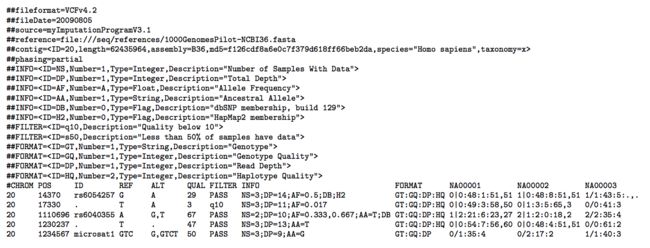
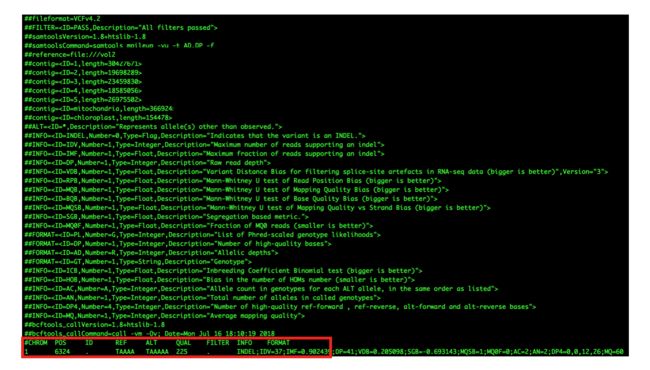
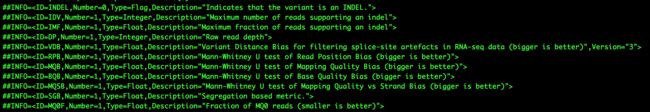
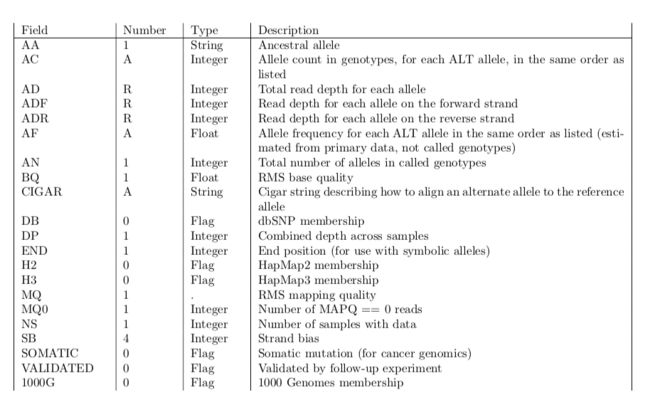
- LocalDateTime 转 String
igotyback
java开发语言
importjava.time.LocalDateTime;importjava.time.format.DateTimeFormatter;publicclassMain{publicstaticvoidmain(String[]args){//获取当前时间LocalDateTimenow=LocalDateTime.now();//定义日期格式化器DateTimeFormatterformat
- 【一起学Rust | 设计模式】习惯语法——使用借用类型作为参数、格式化拼接字符串、构造函数
广龙宇
一起学Rust#Rust设计模式rust设计模式开发语言
提示:文章写完后,目录可以自动生成,如何生成可参考右边的帮助文档文章目录前言一、使用借用类型作为参数二、格式化拼接字符串三、使用构造函数总结前言Rust不是传统的面向对象编程语言,它的所有特性,使其独一无二。因此,学习特定于Rust的设计模式是必要的。本系列文章为作者学习《Rust设计模式》的学习笔记以及自己的见解。因此,本系列文章的结构也与此书的结构相同(后续可能会调成结构),基本上分为三个部分
- 将cmd中命令输出保存为txt文本文件
落难Coder
Windowscmdwindow
最近深度学习本地的训练中我们常常要在命令行中运行自己的代码,无可厚非,我们有必要保存我们的炼丹结果,但是复制命令行输出到txt是非常麻烦的,其实Windows下的命令行为我们提供了相应的操作。其基本的调用格式就是:运行指令>输出到的文件名称或者具体保存路径测试下,我打开cmd并且ping一下百度:pingwww.baidu.com>./data.txt看下相同目录下data.txt的输出:如果你再
- python是什么意思中文-在python中%是什么意思
编程大乐趣
Python中%有两种:1、数值运算:%代表取模,返回除法的余数。如:>>>7%212、%操作符(字符串格式化,stringformatting),说明如下:%[(name)][flags][width].[precision]typecode(name)为命名flags可以有+,-,''或0。+表示右对齐。-表示左对齐。''为一个空格,表示在正数的左侧填充一个空格,从而与负数对齐。0表示使用0填
- 如何部分格式化提示模板:LangChain中的高级技巧
nseejrukjhad
langchainjava服务器python
标题:如何部分格式化提示模板:LangChain中的高级技巧内容:如何部分格式化提示模板:LangChain中的高级技巧引言在使用大型语言模型(LLM)时,提示工程是一个关键环节。LangChain提供了强大的提示模板功能,让我们能更灵活地构建和管理提示。本文将介绍LangChain中一个高级特性-部分格式化提示模板,这个技巧可以让你的提示管理更加高效和灵活。什么是部分格式化提示模板?部分格式化提
- 【目标检测数据集】卡车数据集1073张VOC+YOLO格式
熬夜写代码的平头哥∰
目标检测YOLO人工智能
数据集格式:PascalVOC格式+YOLO格式(不包含分割路径的txt文件,仅仅包含jpg图片以及对应的VOC格式xml文件和yolo格式txt文件)图片数量(jpg文件个数):1073标注数量(xml文件个数):1073标注数量(txt文件个数):1073标注类别数:1标注类别名称:["truck"]每个类别标注的框数:truck框数=1120总框数:1120使用标注工具:labelImg标注
- 番茄西红柿叶子病害分类数据集12882张11类别
futureflsl
数据集分类数据挖掘人工智能
数据集类型:图像分类用,不可用于目标检测无标注文件数据集格式:仅仅包含jpg图片,每个类别文件夹下面存放着对应图片图片数量(jpg文件个数):12882分类类别数:11类别名称:["Bacterial_Spot_Bacteria","Early_Blight_Fungus","Healthy","Late_Blight_Water_Mold","Leaf_Mold_Fungus","Powdery
- 钢筋长度超限检测检数据集VOC+YOLO格式215张1类别
futureflsl
数据集YOLO深度学习机器学习
数据集格式:PascalVOC格式+YOLO格式(不包含分割路径的txt文件,仅仅包含jpg图片以及对应的VOC格式xml文件和yolo格式txt文件)图片数量(jpg文件个数):215标注数量(xml文件个数):215标注数量(txt文件个数):215标注类别数:1标注类别名称:["iron"]每个类别标注的框数:iron框数=215总框数:215使用标注工具:labelImg标注规则:对类别进
- docker
igotyback
eureka云原生
Docker容器的文件系统是隔离的,但是可以通过挂载卷(Volumes)或绑定挂载(BindMounts)将宿主机的文件系统目录映射到容器内部。要查看Docker容器的映射路径,可以使用以下方法:查看容器配置:使用dockerinspect命令可以查看容器的详细配置信息,包括挂载的卷。例如:bashdockerinspect在输出的JSON格式中,查找"Mounts"部分,这里会列出所有的挂载信息
- 关于Mysql 中 Row size too large (> 8126) 错误的解决和理解
秋刀prince
mysqlmysql数据库
提示:啰嗦一嘴,数据库的任何操作和验证前,一定要记得先备份!!!不会有错;文章目录问题发现一、问题导致的可能原因1、页大小2、行格式2.1compact格式2.2Redundant格式2.3Dynamic格式2.4Compressed格式3、BLOB和TEXT列二、解决办法1、修改页大小(不推荐)2、修改行格式3、修改数据类型为BLOB和TEXT列4、其他优化方式(可以参考使用)4.1合理设置数据
- 2024.9.6 Python,华为笔试题总结,字符串格式化,字符串操作,广度优先搜索解决公司组织绩效互评问题,无向图
RaidenQ
python华为leetcode算法力扣广度优先无向图
1.字符串格式化name="Alice"age=30formatted_string="Name:{},Age:{}".format(name,age)print(formatted_string)或者name="Alice"age=30formatted_string=f"Name:{name},Age:{age}"print(formatted_string)2.网络健康检查第一行有两个整数m
- python语法——三目运算符
HappyRocking
pythonpython三目运算符
在java中,有三目运算符,如:intc=(a>b)?a:b表示c取两者中的较大值。但是在python,不能直接这样使用,估计是因为冒号在python有分行的关键作用。那么在python中,如何实现类似功能呢?可以使用ifelse语句,也是一行可以完成,格式为:aifbelsec表示如果b为True,则表达式等于a,否则等于c。如:c=(aif(a>b)elseb)同样是完成了取最大值的功能。
- 第二十 python基础--语句
九樱MOL
目录具体内容1:if语句的使用格式判断语句2:if-else的使用格式3:if-elif-else的使用格式4:if嵌套1:while循环的格式循环语句2:while循环嵌套3:for循环的格式一、判断语句在程序中如果某些条件满足,才能做某件事情,而不满足时不允许做,这就是所谓的判断1.1if语句的使用格式if要判断的条件:条件成立时,要做的事情案例:判断年纪,如果age大于18,输入成年age=
- python批量读取tiff文件_Python Pillow批量转换tif格式到jpg
weixin_39557797
最近因为想要整下网站的壁纸,从网站下载了别人整理好的合集压缩包,解压之后,却发现里面的文件都是tif的,tif格式网站和电脑都不认的,根本不能作壁纸。这时候,就需要转换图片格式了,首先我找了几款转换格式的软件,发现效果都不好,要不是不支持tif格式,要不就是转换出来的图片糊的不行。最终,还是决定用Python的Pillow库来写一个脚本,完成这个任务。下面是整个的小脚本----importosim
- 光盘文件系统 (iso9660) 格式解析
穷人小水滴
光盘文件系统iso9660denoGNU/Linuxjavascript
越简单的系统,越可靠,越不容易出问题.光盘文件系统(iso9660)十分简单,只需不到200行代码,即可实现定位读取其中的文件.参考资料:https://wiki.osdev.org/ISO_9660相关文章:《光盘防水嘛?DVD+R刻录光盘泡水实验》https://blog.csdn.net/secext2022/article/details/140583910《光驱的内部结构及日常使用》ht
- 利用python实现图片格式之间的相互转换
难得北窗高卧
python开发语言
一、概要图片一般有多种格式,常见的图片格式包括:JPEG(.jpg或.jpeg):一种广泛使用的有损压缩格式,适用于摄影图像和网页上的图片。PNG(.png):一种无损压缩格式,支持透明度和更好的图像质量,常用于图标、图形和需要透明背景的图片。该图片是4通道的,外加一个透明通道。如截屏GIF(.gif):一种支持动画和透明度的格式,常用于简单的动画和图标。BMP(.bmp):一种无损格式,存储图像
- Python实现下载当前年份的谷歌影像
sand&wich
python开发语言
在GIS项目和地图应用中,获取最新的地理影像数据是非常重要的。本文将介绍如何使用Python代码从Google地图自动下载当前年份的影像数据,并将其保存为高分辨率的TIFF格式文件。这个过程涉及地理坐标转换、多线程下载和图像处理。关键功能该脚本的核心功能包括:坐标转换:支持WGS-84与WebMercator投影之间转换,以及处理中国GCJ-02偏移。自动化下载:多线程下载地图瓦片,提高效率。图像
- Python实现TIFF 文件转换为 PNG 和 JPG 格式
sand&wich
python开发语言
在日常的图像处理工作中,可能会遇到需要将TIFF格式的图像转换为其他格式的情况,例如PNG和JPG。下面,本文将介绍如何使用Python和GDAL库实现这一功能。准备工作在开始之前,请确保已经安装了必要的库:GDAL(GeospatialDataAbstractionLibrary)可以使用以下命令安装GDAL:pipinstallgdal代码实现以下是一个将TIFF文件转换为PNG文件的示例代码
- COCO 格式的数据集转化为 YOLO 格式的数据集
QYQY77
YOLOpython
"""--json_path输入的json文件路径--save_path保存的文件夹名字,默认为当前目录下的labels。"""importosimportjsonfromtqdmimporttqdmimportargparseparser=argparse.ArgumentParser()parser.add_argument('--json_path',default='./instances
- 非对称加密算法原理与应用2——RSA私钥加密文件
私语茶馆
云部署与开发架构及产品灵感记录RSA2048私钥加密
作者:私语茶馆1.相关章节(1)非对称加密算法原理与应用1——秘钥的生成-CSDN博客第一章节讲述的是创建秘钥对,并将公钥和私钥导出为文件格式存储。本章节继续讲如何利用私钥加密内容,包括从密钥库或文件中读取私钥,并用RSA算法加密文件和String。2.私钥加密的概述本文主要基于第一章节的RSA2048bit的非对称加密算法讲述如何利用私钥加密文件。这种加密后的文件,只能由该私钥对应的公钥来解密。
- 4款毕业论文参考文献格式生成器(附加详细步骤)
小猪包333
写论文人工智能深度学习计算机视觉AI写作
在撰写毕业论文时,参考文献的格式规范是至关重要的。为了帮助学生和学者们更高效地生成符合要求的参考文献格式,本文将详细介绍四款推荐的参考文献格式生成器,并提供详细的使用步骤。1.千笔-AIPassPaper千笔-AIPassPaper是一款先进的AI辅助论文写作工具,不仅能够自动生成大纲、开题报告,还能一键生成参考文献。AI论文,免费大纲,10分钟3万字https://www.aipaperpass
- Linux CTF逆向入门
蚁景网络安全
linux运维CTF
1.ELF格式我们先来看看ELF文件头,如果想详细了解,可以查看ELF的manpage文档。关于ELF更详细的说明:e_shoff:节头表的文件偏移量(字节)。如果文件没有节头表,则此成员值为零。sh_offset:表示了该section(节)离开文件头部位置的距离+-------------------+|ELFheader|---++--------->+-------------------
- 【Golang】实现 Excel 文件下载功能
RumIV
Golanggolangexcel开发语言
在当今的网络应用开发中,提供数据导出功能是一项常见的需求。Excel作为一种广泛使用的电子表格格式,通常是数据导出的首选格式之一。在本教程中,我们将学习如何使用Go语言和GinWeb框架来创建一个Excel文件,并允许用户通过HTTP请求下载该文件。准备工作在开始之前,请确保您的开发环境中已经安装了Go语言和相关的开发工具。此外,您还需要安装GinWeb框架和excelize包,这两个包都将用于我
- Shell脚本中sed使用
jcrhl321
linux
目录一、sed编辑器1、sed概述2、sed的工作流程3、sed命令的常见格式4、sed命令常用操作二、sed常用命令使用1、sed打印2、sed删除3、sed替换4、sed插入与增加4、sed剪切粘贴与复制粘贴一、sed编辑器sed(StreamEDitor)是一个强大而简单的文本解析转换工具,可以读取文本,并根据指定的条件对文本内容进行编辑(删除、替换、添加、移动等),最后输出所有行或者仅输出
- [数据集][目标检测]汽车头部尾部检测数据集VOC+YOLO格式5319张3类别
FL1623863129
数据集目标检测汽车YOLO
数据集制作单位:未来自主研究中心(FIRC)版权单位:未来自主研究中心(FIRC)版权声明:数据集仅仅供个人使用,不得在未授权情况下挂淘宝、咸鱼等交易网站公开售卖,由此引发的法律责任需自行承担数据集格式:PascalVOC格式+YOLO格式(不包含分割路径的txt文件,仅仅包含jpg图片以及对应的VOC格式xml文件和yolo格式txt文件)图片数量(jpg文件个数):5319标注数量(xml文件
- 菜鸟教你修U盘
zoyation
文章工具百度google杀毒软件
优盘是好多人都在使用的便携设备在给我们带来方便的同时,也带来困惑由于这样或那样的原因,优盘总是会出这样或那样的毛病小的毛病是中了一般的病毒,用杀毒软件就能清除,一般不会有什么损失稍微大的毛病是由于使用不当(拔插优盘时没有安全弹出等原因)导致再次使用时优盘不能打开、不能拷贝文件、不能格式化、提示写保护或者使用时要等很久才显示出来盘符并且出现一种卡机的状态再大点的毛病就是根本在电脑上就显示不出来盘符(
- ffmpeg批量将tif文件转成jpeg格式
winfredzhang
图像工具ffmpegtifjpeg转换
1、cmd2、切换到安装ffmpeg的路径。3、输入命令:ffmpeg-start_number001-i"D:\ocr\%03d.tif"-start_number001-pix_fmtyuv420p-qscale:v1"D:\ocr\%03d.jpg"结果。
- 使用python抽取post接口数据示例
中台小A
pythonpython开发语言
postman调用接口post接口https://inner-XXXXX.XXXXX.com/wXX/api/XXXXXctoryLake?user_key=XXXXXXXXXXXX,在boday的row里输入Jason格式的{"wasStartDay":"2024-09-03"}importrequestsurl='https://inner-XXXXX.XXXXX.com/wXX/api/XX
- 导致格式错误的 Lambda 代理响应的原因以及如何修复它
zqhdz米时空
汇编
当人们尝试使用AWSAPIGateway和AWSLambda构建无服务器应用程序时,经常出现的一个问题是_由于配置错误而执行失败:Lambda代理响应格式错误。_没有什么比通用错误消息更糟糕的了,它们不会告诉您解决问题所需的任何内容,对吧?AWS并不是以其错误消息设计而闻名,如果甚至可以这样称呼它的话,更不用说为您提供解决问题的方法了。那么如何修复这个Lambda错误以及是什么原因造成的呢?花椒壳
- 【Golang】使用 Golang 语言和 excelize 库将数据写入Excel
不爱洗脚的小滕
golangexcel开发语言
文章目录前言一、Excelize简介二、代码实现1.获取依赖2.示例代码三、总结前言在数据处理和分析中,Excel作为一种常见的电子表格格式,被广泛应用于各种场景。然而,如何在Go语言中有效地处理Excel文件呢?在这篇博客中,我将介绍如何使用Go语言和excelize库将数据写入Excel文件。一、Excelize简介Excelize是一个用于读取和写入MicrosoftExcel™(XLSX)
- Java开发中,spring mvc 的线程怎么调用?
小麦麦子
springmvc
今天逛知乎,看到最近很多人都在问spring mvc 的线程http://www.maiziedu.com/course/java/ 的启动问题,觉得挺有意思的,那哥们儿问的也听仔细,下面的回答也很详尽,分享出来,希望遇对遇到类似问题的Java开发程序猿有所帮助。
问题:
在用spring mvc架构的网站上,设一线程在虚拟机启动时运行,线程里有一全局
- maven依赖范围
bitcarter
maven
1.test 测试的时候才会依赖,编译和打包不依赖,如junit不被打包
2.compile 只有编译和打包时才会依赖
3.provided 编译和测试的时候依赖,打包不依赖,如:tomcat的一些公用jar包
4.runtime 运行时依赖,编译不依赖
5.默认compile
依赖范围compile是支持传递的,test不支持传递
1.传递的意思是项目A,引用
- Jaxb org.xml.sax.saxparseexception : premature end of file
darrenzhu
xmlprematureJAXB
如果在使用JAXB把xml文件unmarshal成vo(XSD自动生成的vo)时碰到如下错误:
org.xml.sax.saxparseexception : premature end of file
很有可能时你直接读取文件为inputstream,然后将inputstream作为构建unmarshal需要的source参数。InputSource inputSource = new In
- CSS Specificity
周凡杨
html权重Specificitycss
有时候对于页面元素设置了样式,可为什么页面的显示没有匹配上呢? because specificity
CSS 的选择符是有权重的,当不同的选择符的样式设置有冲突时,浏览器会采用权重高的选择符设置的样式。
规则:
HTML标签的权重是1
Class 的权重是10
Id 的权重是100
- java与servlet
g21121
servlet
servlet 搞java web开发的人一定不会陌生,而且大家还会时常用到它。
下面是java官方网站上对servlet的介绍: java官网对于servlet的解释 写道
Java Servlet Technology Overview Servlets are the Java platform technology of choice for extending and enha
- eclipse中安装maven插件
510888780
eclipsemaven
1.首先去官网下载 Maven:
http://www.apache.org/dyn/closer.cgi/maven/binaries/apache-maven-3.2.3-bin.tar.gz
下载完成之后将其解压,
我将解压后的文件夹:apache-maven-3.2.3,
并将它放在 D:\tools目录下,
即 maven 最终的路径是:D:\tools\apache-mave
- jpa@OneToOne关联关系
布衣凌宇
jpa
Nruser里的pruserid关联到Pruser的主键id,实现对一个表的增删改,另一个表的数据随之增删改。
Nruser实体类
//*****************************************************************
@Entity
@Table(name="nruser")
@DynamicInsert @Dynam
- 我的spring学习笔记11-Spring中关于声明式事务的配置
aijuans
spring事务配置
这两天学到事务管理这一块,结合到之前的terasoluna框架,觉得书本上讲的还是简单阿。我就把我从书本上学到的再结合实际的项目以及网上看到的一些内容,对声明式事务管理做个整理吧。我看得Spring in Action第二版中只提到了用TransactionProxyFactoryBean和<tx:advice/>,定义注释驱动这三种,我承认后两种的内容很好,很强大。但是实际的项目当中
- java 动态代理简单实现
antlove
javahandlerproxydynamicservice
dynamicproxy.service.HelloService
package dynamicproxy.service;
public interface HelloService {
public void sayHello();
}
dynamicproxy.service.impl.HelloServiceImpl
package dynamicp
- JDBC连接数据库
百合不是茶
JDBC编程JAVA操作oracle数据库
如果我们要想连接oracle公司的数据库,就要首先下载oralce公司的驱动程序,将这个驱动程序的jar包导入到我们工程中;
JDBC链接数据库的代码和固定写法;
1,加载oracle数据库的驱动;
&nb
- 单例模式中的多线程分析
bijian1013
javathread多线程java多线程
谈到单例模式,我们立马会想到饿汉式和懒汉式加载,所谓饿汉式就是在创建类时就创建好了实例,懒汉式在获取实例时才去创建实例,即延迟加载。
饿汉式:
package com.bijian.study;
public class Singleton {
private Singleton() {
}
// 注意这是private 只供内部调用
private static
- javascript读取和修改原型特别需要注意原型的读写不具有对等性
bijian1013
JavaScriptprototype
对于从原型对象继承而来的成员,其读和写具有内在的不对等性。比如有一个对象A,假设它的原型对象是B,B的原型对象是null。如果我们需要读取A对象的name属性值,那么JS会优先在A中查找,如果找到了name属性那么就返回;如果A中没有name属性,那么就到原型B中查找name,如果找到了就返回;如果原型B中也没有
- 【持久化框架MyBatis3六】MyBatis3集成第三方DataSource
bit1129
dataSource
MyBatis内置了数据源的支持,如:
<environments default="development">
<environment id="development">
<transactionManager type="JDBC" />
<data
- 我程序中用到的urldecode和base64decode,MD5
bitcarter
cMD5base64decodeurldecode
这里是base64decode和urldecode,Md5在附件中。因为我是在后台所以需要解码:
string Base64Decode(const char* Data,int DataByte,int& OutByte)
{
//解码表
const char DecodeTable[] =
{
0, 0, 0, 0, 0, 0
- 腾讯资深运维专家周小军:QQ与微信架构的惊天秘密
ronin47
社交领域一直是互联网创业的大热门,从PC到移动端,从OICQ、MSN到QQ。到了移动互联网时代,社交领域应用开始彻底爆发,直奔黄金期。腾讯在过去几年里,社交平台更是火到爆,QQ和微信坐拥几亿的粉丝,QQ空间和朋友圈各种刷屏,写心得,晒照片,秀视频,那么谁来为企鹅保驾护航呢?支撑QQ和微信海量数据背后的架构又有哪些惊天内幕呢?本期大讲堂的内容来自今年2月份ChinaUnix对腾讯社交网络运营服务中心
- java-69-旋转数组的最小元素。把一个数组最开始的若干个元素搬到数组的末尾,我们称之为数组的旋转。输入一个排好序的数组的一个旋转,输出旋转数组的最小元素
bylijinnan
java
public class MinOfShiftedArray {
/**
* Q69 旋转数组的最小元素
* 把一个数组最开始的若干个元素搬到数组的末尾,我们称之为数组的旋转。输入一个排好序的数组的一个旋转,输出旋转数组的最小元素。
* 例如数组{3, 4, 5, 1, 2}为{1, 2, 3, 4, 5}的一个旋转,该数组的最小值为1。
*/
publ
- 看博客,应该是有方向的
Cb123456
反省看博客
看博客,应该是有方向的:
我现在就复习以前的,在补补以前不会的,现在还不会的,同时完善完善项目,也看看别人的博客.
我刚突然想到的:
1.应该看计算机组成原理,数据结构,一些算法,还有关于android,java的。
2.对于我,也快大四了,看一些职业规划的,以及一些学习的经验,看看别人的工作总结的.
为什么要写
- [开源与商业]做开源项目的人生活上一定要朴素,尽量减少对官方和商业体系的依赖
comsci
开源项目
为什么这样说呢? 因为科学和技术的发展有时候需要一个平缓和长期的积累过程,但是行政和商业体系本身充满各种不稳定性和不确定性,如果你希望长期从事某个科研项目,但是却又必须依赖于某种行政和商业体系,那其中的过程必定充满各种风险。。。
所以,为避免这种不确定性风险,我
- 一个 sql优化 ([精华] 一个查询优化的分析调整全过程!很值得一看 )
cwqcwqmax9
sql
见 http://www.itpub.net/forum.php?mod=viewthread&tid=239011
Web翻页优化实例
提交时间: 2004-6-18 15:37:49 回复 发消息
环境:
Linux ve
- Hibernat and Ibatis
dashuaifu
Hibernateibatis
Hibernate VS iBATIS 简介 Hibernate 是当前最流行的O/R mapping框架,当前版本是3.05。它出身于sf.net,现在已经成为Jboss的一部分了 iBATIS 是另外一种优秀的O/R mapping框架,当前版本是2.0。目前属于apache的一个子项目了。 相对Hibernate“O/R”而言,iBATIS 是一种“Sql Mappi
- 备份MYSQL脚本
dcj3sjt126com
mysql
#!/bin/sh
# this shell to backup mysql
#
[email protected] (QQ:1413161683 DuChengJiu)
_dbDir=/var/lib/mysql/
_today=`date +%w`
_bakDir=/usr/backup/$_today
[ ! -d $_bakDir ] && mkdir -p
- iOS第三方开源库的吐槽和备忘
dcj3sjt126com
ios
转自
ibireme的博客 做iOS开发总会接触到一些第三方库,这里整理一下,做一些吐槽。 目前比较活跃的社区仍旧是Github,除此以外也有一些不错的库散落在Google Code、SourceForge等地方。由于Github社区太过主流,这里主要介绍一下Github里面流行的iOS库。 首先整理了一份
Github上排名靠
- html wlwmanifest.xml
eoems
htmlxml
所谓优化wp_head()就是把从wp_head中移除不需要元素,同时也可以加快速度。
步骤:
加入到function.php
remove_action('wp_head', 'wp_generator');
//wp-generator移除wordpress的版本号,本身blog的版本号没什么意义,但是如果让恶意玩家看到,可能会用官网公布的漏洞攻击blog
remov
- 浅谈Java定时器发展
hacksin
java并发timer定时器
java在jdk1.3中推出了定时器类Timer,而后在jdk1.5后由Dou Lea从新开发出了支持多线程的ScheduleThreadPoolExecutor,从后者的表现来看,可以考虑完全替代Timer了。
Timer与ScheduleThreadPoolExecutor对比:
1.
Timer始于jdk1.3,其原理是利用一个TimerTask数组当作队列
- 移动端页面侧边导航滑入效果
ini
jqueryWebhtml5cssjavascirpt
效果体验:http://hovertree.com/texiao/mobile/2.htm可以使用移动设备浏览器查看效果。效果使用到jquery-2.1.4.min.js,该版本的jQuery库是用于支持HTML5的浏览器上,不再兼容IE8以前的浏览器,现在移动端浏览器一般都支持HTML5,所以使用该jQuery没问题。HTML文件代码:
<!DOCTYPE html>
<h
- AspectJ+Javasist记录日志
kane_xie
aspectjjavasist
在项目中碰到这样一个需求,对一个服务类的每一个方法,在方法开始和结束的时候分别记录一条日志,内容包括方法名,参数名+参数值以及方法执行的时间。
@Override
public String get(String key) {
// long start = System.currentTimeMillis();
// System.out.println("Be
- redis学习笔记
MJC410621
redisNoSQL
1)nosql数据库主要由以下特点:非关系型的、分布式的、开源的、水平可扩展的。
1,处理超大量的数据
2,运行在便宜的PC服务器集群上,
3,击碎了性能瓶颈。
1)对数据高并发读写。
2)对海量数据的高效率存储和访问。
3)对数据的高扩展性和高可用性。
redis支持的类型:
Sring 类型
set name lijie
get name lijie
set na
- 使用redis实现分布式锁
qifeifei
在多节点的系统中,如何实现分布式锁机制,其中用redis来实现是很好的方法之一,我们先来看一下jedis包中,有个类名BinaryJedis,它有个方法如下:
public Long setnx(final byte[] key, final byte[] value) {
checkIsInMulti();
client.setnx(key, value);
ret
- BI并非万能,中层业务管理报表要另辟蹊径
张老师的菜
大数据BI商业智能信息化
BI是商业智能的缩写,是可以帮助企业做出明智的业务经营决策的工具,其数据来源于各个业务系统,如ERP、CRM、SCM、进销存、HER、OA等。
BI系统不同于传统的管理信息系统,他号称是一个整体应用的解决方案,是融入管理思想的强大系统:有着系统整体的设计思想,支持对所有
- 安装rvm后出现rvm not a function 或者ruby -v后提示没安装ruby的问题
wudixiaotie
function
1.在~/.bashrc最后加入
[[ -s "$HOME/.rvm/scripts/rvm" ]] && source "$HOME/.rvm/scripts/rvm"
2.重新启动terminal输入:
rvm use ruby-2.2.1 --default
把当前安装的ruby版本设为默
 0_0.png
0_0.png 0_1.png
0_1.png