花花写于2020-01-29 这个仍然是TCGA课程里的内容,但图是箱线图的美化版,可以用在任何领域,不局限于TCGA。
0.输入数据
rm(list=ls())
load(file = "for_boxplot.Rdata")
这里面有三个数据:
expr和meta是miRNA的表达矩阵和临床信息,由GDC下载整理得到。
mut是突变信息,读取maf得到的数据框筛选了几列得到。
在生信星球公众号回复“box”即可获得。也可参照前面的笔记自己获得。
1.比较任意miRNA在tumor和normal样本中的表达量
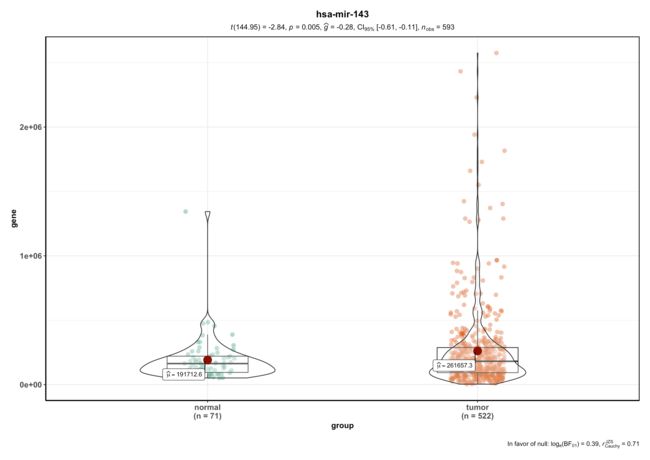
这个只需要表达矩阵,以hsa-mir-143为例画图,可替换为其他任意miRNA。
expr[1:4,1:4]
#> TCGA-A3-3307-01A-01T-0860-13 TCGA-A3-3308-01A-02R-1324-13
#> hsa-let-7a-1 5056 14503
#> hsa-let-7a-2 10323 29238
#> hsa-let-7a-3 5429 14738
#> hsa-let-7b 17908 37062
#> TCGA-A3-3311-01A-02R-1324-13 TCGA-A3-3313-01A-02R-1324-13
#> hsa-let-7a-1 8147 7138
#> hsa-let-7a-2 16325 14356
#> hsa-let-7a-3 8249 7002
#> hsa-let-7b 28984 6909
group_list=ifelse(as.numeric(substr(colnames(expr),14,15)) < 10,'tumor','normal')
table(group_list)
#> group_list
#> normal tumor
#> 71 522
library(ggstatsplot)
dat = data.frame(gene = expr["hsa-mir-143",],
group = group_list)
ggbetweenstats(data = dat, x = group, y = gene,title = "hsa-mir-143")
#> Note: Shapiro-Wilk Normality Test for gene: p-value = < 0.001
#>
#> Note: Bartlett's test for homogeneity of variances for factor group: p-value = < 0.001
#>
2.任意miRNA在任意两个分组中的表达量对比
只要肿瘤样本,522个,只要是可以根据临床信息查到或得到的分组,例如生死、人种、阶段,都可以拿来做分组。
需要注意调整样本顺序,一一对应。
expf = expr[,as.numeric(substr(colnames(expr),14,15)) < 10]
library(stringr)
x1=str_sub(colnames(expf),1,12)
x2=str_to_upper(meta$patient.bcr_patient_barcode)
table(x2 %in% x1)
#>
#> FALSE TRUE
#> 21 516
table(x1 %in% x2)
#>
#> TRUE
#> 522
length(unique(x1))
#> [1] 516
#发现一个问题,样本的前12位代表病人的编号,列名是有重复的,为了一对一关系,去重复走起
expf = expf[,!duplicated(str_sub(colnames(expf),1,12))]
x1=str_sub(colnames(expf),1,12)
x2=str_to_upper(meta$patient.bcr_patient_barcode)
table(x2 %in% x1)
#>
#> FALSE TRUE
#> 21 516
meta = meta[x2 %in% x1 ,]
#按照生死、人种、阶段分组看看
table(meta$patient.vital_status)
#>
#> alive dead
#> 358 158
table(meta$patient.stage_event.pathologic_stage)
#>
#> stage i stage ii stage iii stage iv
#> 254 55 124 83
table(meta$patient.race)
#>
#> asian black or african american
#> 8 56
#> white
#> 445
dat = data.frame(gene = expf["hsa-mir-143",],
vital_status = meta$patient.vital_status,
stage = meta$patient.stage_event.pathologic_stage,
race = meta$patient.race)
p1 = ggbetweenstats(data = dat, x = vital_status, y = gene,title = "hsa-mir-143")
#> Note: Shapiro-Wilk Normality Test for gene: p-value = < 0.001
#>
#> Note: Bartlett's test for homogeneity of variances for factor vital_status: p-value = 0.101
#>
p2 = ggbetweenstats(data = dat, x = stage, y = gene,title = "hsa-mir-143")
#> Note: 95% CI for effect size estimate was computed with 100 bootstrap samples.
#>
#> Note: Shapiro-Wilk Normality Test for gene: p-value = < 0.001
#>
#> Note: Bartlett's test for homogeneity of variances for factor stage: p-value = < 0.001
#>
p3 = ggbetweenstats(data = dat, x = race, y = gene,title = "hsa-mir-143")
#> Note: 95% CI for effect size estimate was computed with 100 bootstrap samples.
#>
#> Note: Shapiro-Wilk Normality Test for gene: p-value = < 0.001
#>
#> Note: Bartlett's test for homogeneity of variances for factor race: p-value = 0.001
#>
library(patchwork)
p1+p2+p3

3.根据某个基因是否突变分组比较某miRNA的表达量
dim(expf)
#> [1] 552 516
head(mut)
#> Hugo_Symbol Chromosome Start_Position Tumor_Sample_Barcode
#> 1: HNRNPCL2 chr1 13115853 TCGA-G6-A8L7-01A-11D-A36X-10
#> 2: ERMAP chr1 42842993 TCGA-G6-A8L7-01A-11D-A36X-10
#> 3: FAAH chr1 46394349 TCGA-G6-A8L7-01A-11D-A36X-10
#> 4: EPS15 chr1 51448116 TCGA-G6-A8L7-01A-11D-A36X-10
#> 5: HMGCS2 chr1 119764248 TCGA-G6-A8L7-01A-11D-A36X-10
#> 6: NOS1AP chr1 162367063 TCGA-G6-A8L7-01A-11D-A36X-10
#> t_vaf pos
#> 1: 0.2148148 chr1:13115853
#> 2: 0.1650165 chr1:42842993
#> 3: 0.3114754 chr1:46394349
#> 4: 0.1677852 chr1:51448116
#> 5: 0.2539683 chr1:119764248
#> 6: 0.2098765 chr1:162367063
length(unique(str_sub(mut$Tumor_Sample_Barcode,1,12)))
#> [1] 336
table(x1 %in% unique(str_sub(mut$Tumor_Sample_Barcode,1,12)))
#>
#> FALSE TRUE
#> 185 331
#522个样本中有331个有突变信息记录,将这些样本对应的表达矩阵取出来。
expm = expf[,x1 %in% unique(str_sub(mut$Tumor_Sample_Barcode,1,12))]
library(dplyr)
VHL_mut = mut %>%
filter(Hugo_Symbol=='VHL') %>%
as.data.frame() %>%
pull(Tumor_Sample_Barcode) %>%
as.character() %>%
substr(1,12)
#false 对应的是未突变样本,true是突变样本
tail(rownames(expm))
#> [1] "hsa-mir-944" "hsa-mir-95" "hsa-mir-96" "hsa-mir-98" "hsa-mir-99a"
#> [6] "hsa-mir-99b"
dat=data.frame(gene=log2(expm['hsa-mir-98',]),
mut= substr(colnames(expm),1,12) %in% VHL_mut)
ggbetweenstats(data = dat, x = mut, y = gene)
#> Note: Shapiro-Wilk Normality Test for gene: p-value = 0.159
#>
#> Note: Bartlett's test for homogeneity of variances for factor mut: p-value = 0.031
#>
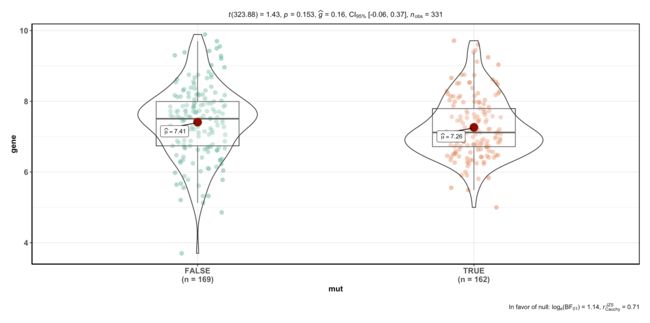
可以看到,自己分组后挑选任意基因,p值可能大可能小,如果不想直接做可视化,也可先批量计算p值,然后挑选p<0.05的基因进行可视化。
res.aov <- aov(gene ~ as.factor(mut), data = dat)
summary(res.aov)
#> Df Sum Sq Mean Sq F value Pr(>F)
#> as.factor(mut) 1 1.81 1.809 2.035 0.155
#> Residuals 329 292.47 0.889
TukeyHSD(res.aov)
#> Tukey multiple comparisons of means
#> 95% family-wise confidence level
#>
#> Fit: aov(formula = gene ~ as.factor(mut), data = dat)
#>
#> $`as.factor(mut)`
#> diff lwr upr p adj
#> TRUE-FALSE -0.1479028 -0.3518444 0.05603875 0.1546273
summary(res.aov)[[1]]$`Pr(>F)`[1]
#> [1] 0.1546273
这样的会,批量计算也很简单了,用apply即可。
微信公众号生信星球同步更新我的文章,欢迎大家扫码关注!

我们有为生信初学者准备的学习小组, 点击查看◀️
想要参加我的线上线下课程,也可加好友咨询
如果需要提问,请先看 生信星球答疑公告