生信人的linux考试
http://www.bio-info-trainee.com/2900.html
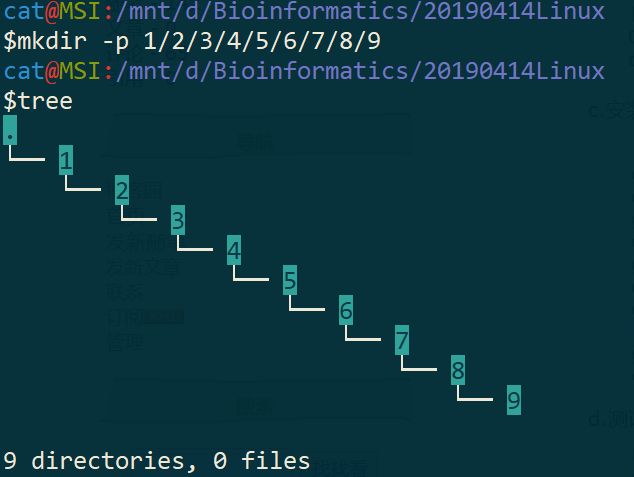
一、在任意文件夹下面创建形如 1/2/3/4/5/6/7/8/9 格式的文件夹系列。
$ mkdir -p 1/2/3/4/5/6/7/8/9
$ tree
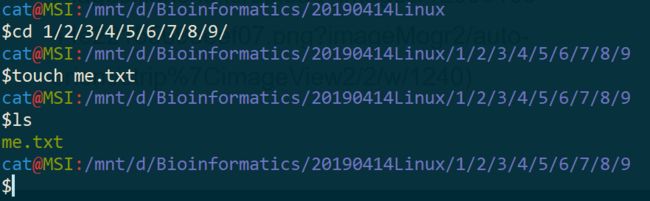
二、在创建好的文件夹下面,比如我的是 /Users/jimmy/tmp/1/2/3/4/5/6/7/8/9 ,里面创建文本文件 me.txt
$cd 1/2/3/4/5/6/7/8/9
$touch me.txt
$ls
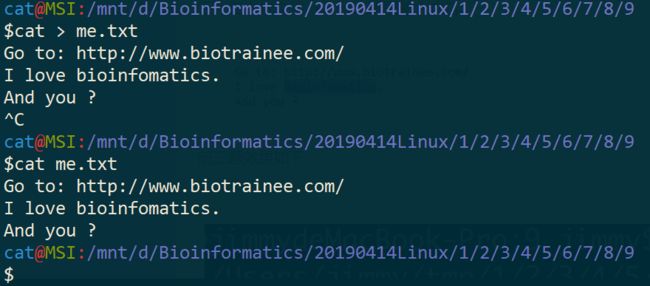
三、在文本文件 me.txt 里面输入内容:
Go to: http://www.biotrainee.com/
I love bioinfomatics.
And you ?
$cat > me.txt
Go to: http://www.biotrainee.com/
I love bioinfomatics.
And you ?
^C
$cat me.txt
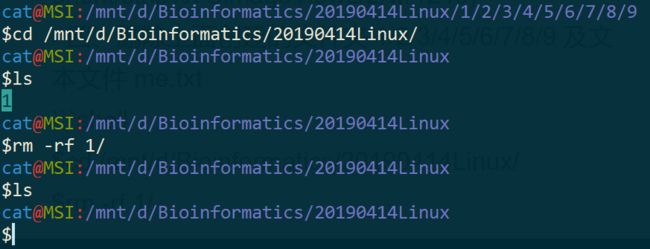
四、删除上面创建的文件夹 1/2/3/4/5/6/7/8/9 及文本文件 me.txt
$cd /mnt/d/Bioinformatics/20190414Linux/
$rm -rf 1/
$ls
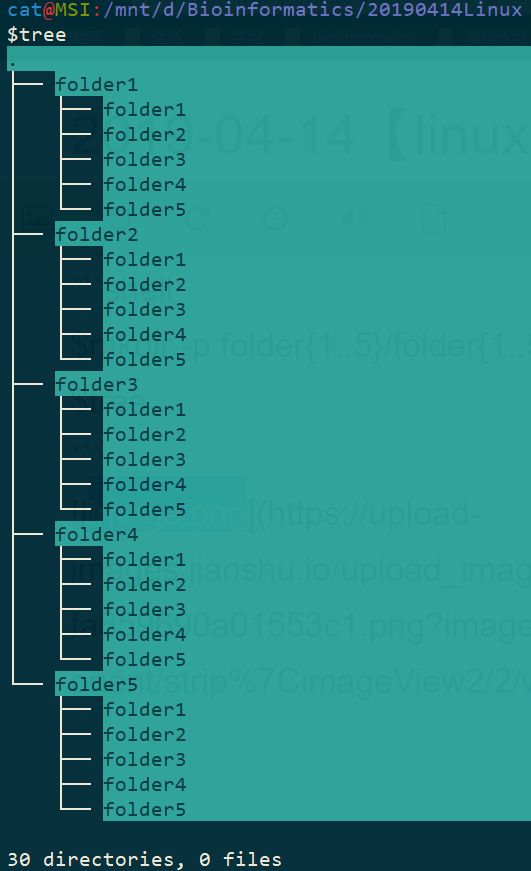
五、在任意文件夹下面创建 folder1~5这5个文件夹,然后每个文件夹下面继续创建 folder1~5这5个文件夹,效果如下:
$mkdir -p folder{1..5}/folder{1..5}
$tree

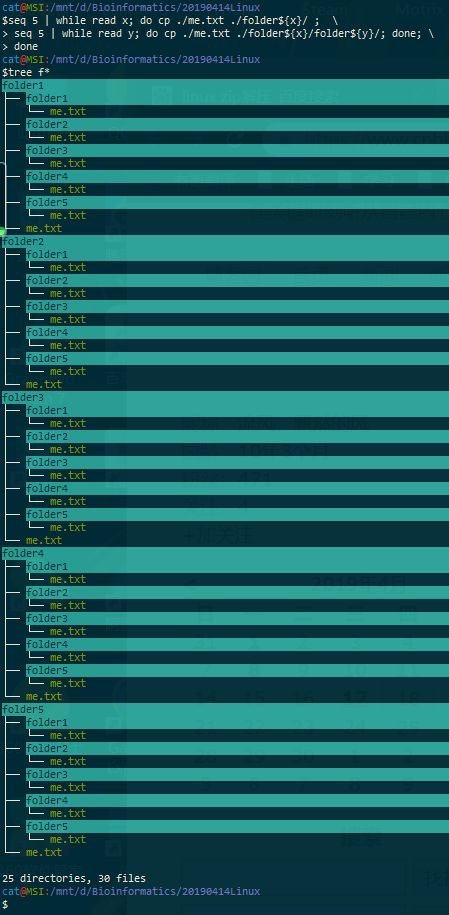
六、在第五题创建的每一个文件夹下面都 创建第二题文本文件 me.txt ,内容也要一样。(这个题目难度超纲,建议一个月后再回过头来做)
$cat > me.txt
Go to: http://www.biotrainee.com/
I love bioinfomatics.
And you ?
^C
$seq 5 | while read x; do cp ./me.txt ./folder${x}/ ; \
>seq 5 | while read y; do cp ./me.txt ./folder${x}/folder${y}/; done; \
>done
$tree f*
七,再次删除掉前面几个步骤建立的文件夹及文件
$rm -rf *
$ls
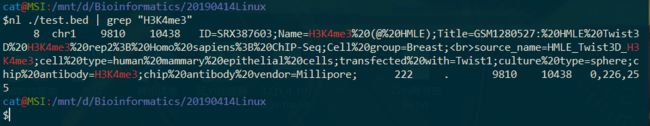
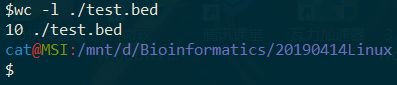
八、下载 http://www.biotrainee.com/jmzeng/igv/test.bed 文件,后在里面选择含有 H3K4me3 的那一行是第几行,该文件总共有几行。
$wget -c http://www.biotrainee.com/jmzeng/igv/test.bed
$nl ./test.bed | grep "H3K4me3"

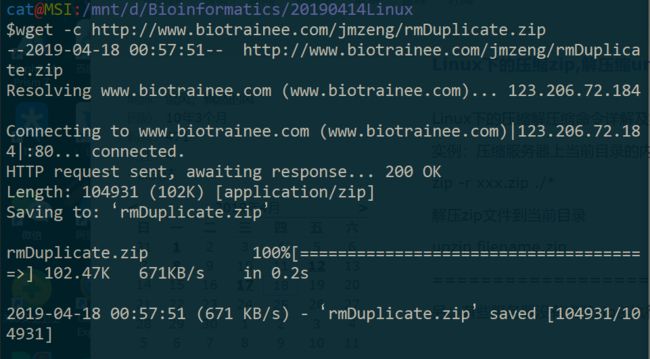
九、下载 http://www.biotrainee.com/jmzeng/rmDuplicate.zip 文件,并且解压,查看里面的文件夹结构
$wget -c http://www.biotrainee.com/jmzeng/rmDuplicate.zip
$unzip rmDuplicate.zip
$tree

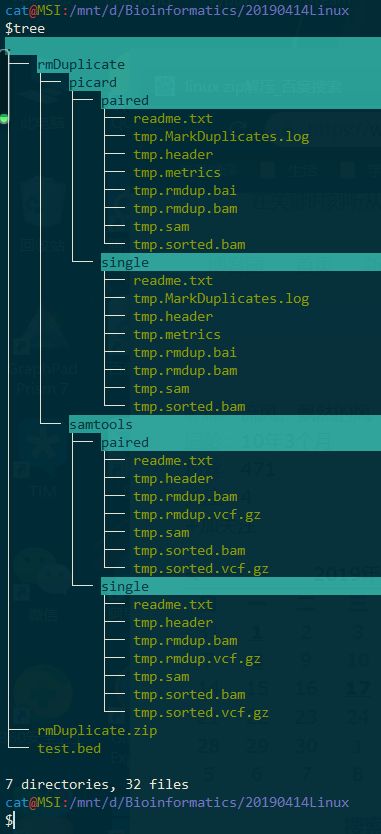
十、打开第九题解压的文件,进入 rmDuplicate/samtools/single 文件夹里面,查看后缀为 .sam 的文件,搞清楚 生物信息学里面的SAM/BAM 定义是什么。
$cd rmDuplicate/samtools/single/
$ls
$head -1 tmp.sam |less -S
SRR1042600.42157053 0 chr1 629895 42 51M * 0 0 ATAACCAATACTACCAATCANTACTCATCATTAATAATCATAATGGCTATA CCCFFFFFHHHHHJJJIIJJ#3AGIIJJJIJJDGIGJJIHIIJJJJIJHII AS:i:-6 XN:i:0 XM:i:2 XO:i:0 XG:i:0 NM:i:2 MD:Z:6C13C30 YT:Z:UU
SAM的全称是sequence alignment/map format。而BAM就是SAM的二进制文件,也就是压缩格式的sam文件。
在SAM输出的结果中每一行都包括十二项通过Tab分隔(\t),从左到右分别是:
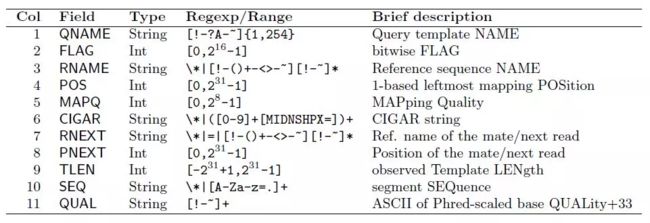
QNAME 表示reads名称;
-
FLAG:表示比对的结果,由数字表示,不同的数值含义不同,其列表如下:
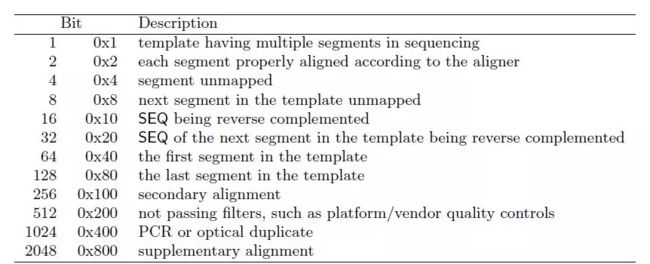
RNAME:表示参考序列的名称,如基因组的染色体编号等,如果没有比对上则显示为*;
POS:表示比对的起始位置,以1开始计数,如果没有比对上则显示为0;
MAPQ:比对质量;(数字越大,特异性越高)
CIGAR:字符串,即比对的详细情况, 记录插入,缺失,错配,后剪切拼接的接头;
RNEXT:双末端测序中下一个reads比对的参考系列的名称,如果没有则用 " * " 表示,如果和前一个reads比对到同一个参考序列则用" = "表示;
PNEXT:下一个reads比对到参考序列上的位置,如果没有则用0表示;
TLEN:序列模板的长度;
SEQ:reads的序列信息;
QUAL:reads的序列质量信息;
可选字段:格式如:TAG:TYPE:VALUE,其中TAG有两个大写字母组成,每个TAG代表一类信息,每一行一个TAG只能出现一次,TYPE表示TAG对应值的类型,可以是字符串、整数、字节、数组等。
【参考】
SAM格式简介
SAM格式详解
十一、安装 samtools 软件
1.搜索samtools,找到官网http://www.htslib.org/download/
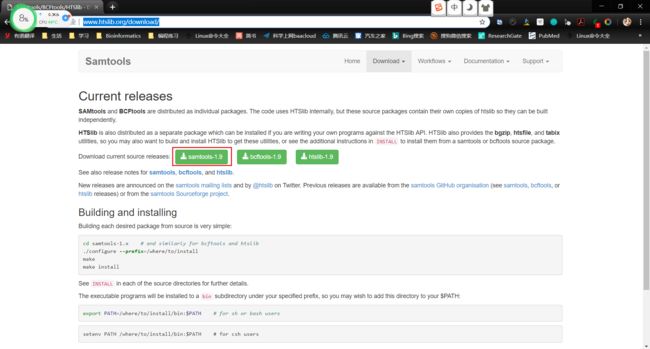
- 右键点击
samtools-1.9,复制链接地址https://github.com/samtools/samtools/releases/download/1.9/samtools-1.9.tar.bz2
 复制下载地址
复制下载地址 - 复制到linux终端,下载文件;
$wget -c https://github.com/samtools/samtools/releases/download/1.9/samtools-1.9.tar.bz2
-
按照官网protocol,安装软件。
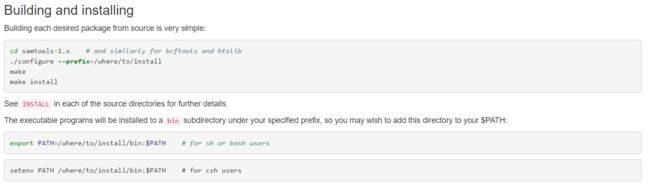 官网protocol
官网protocol
【坑1】./configure时多次失败,按照提示安装各种配套程序,最后./configure通过,make,make install,完成安装。
$sudo apt-get install libncurses5-dev
$sudo apt-get install zlib1g-dev
$sudo apt-get install libbz2-dev
$sudo apt-get install liblzma-dev
【坑2】系统问题,win10系统Linux子系统是没有独立的环境变量PATH的,所以,按照protocol把samtools的文件夹加入到PATH以后,疯狂报错,连ls命令都没法用了,只好重装子系统。
解决方案:用alias或者全路径调用samtools
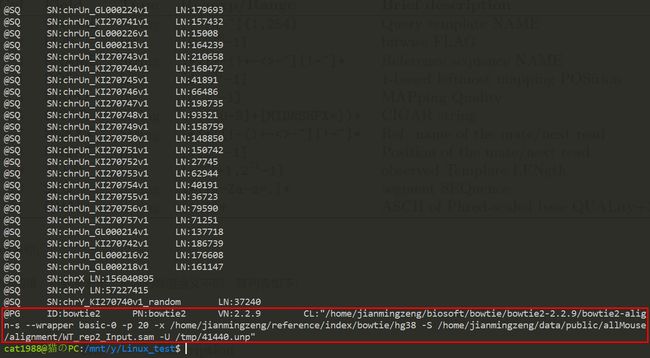
十二、打开 后缀为BAM 的文件,找到产生该文件的命令。 提示一下命令是:
/home/jianmingzeng/biosoft/bowtie/bowtie2-2.2.9/bowtie2-align-s --wrapper basic-0 -p 20 -x /home/jianmingzeng/reference/index/bowtie/hg38 -S /home/jianmingzeng/data/public/allMouse/alignment/WT_rep2_Input.sam -U /tmp/41440.unp
$samtools view tmp.rmdup.bam | less -SN

但是并没有找到相关命令行╮(╯▽╰)╭
偷看了别人的答案,samtools view需要加参数-H
产生该文件的命令存在于表头信息中,而samtools view默认是不输出表头的。
参数-H是只显示文件的表头信息。
再次运行
$samtools view -H tmp.sorted.bam
[SAMtools] 常用指令总结
十三题、根据上面的命令,找到我使用的参考基因组
/home/jianmingzeng/reference/index/bowtie/hg38具体有多少条染色体。
十四题、上面的后缀为
BAM的文件的第二列,只有 0 和 16 两个数字,用cut/sort/uniq等命令统计它们的个数。
$ls *.bam
tmp.rmdup.bam tmp.sorted.bam
$samtools view tmp.rmdup.bam | cut -f 2 | sort | uniq -c
16 0
12 16
$samtools view tmp.sorted.bam | cut -f 2 | sort | uniq -c
29 0
24 16
十五题、重新打开
rmDuplicate/samtools/paired文件夹下面的后缀为BAM的文件,再次查看第二列,并且统计
$cd ../paired/
$ls *.bam
tmp.rmdup.bam tmp.sorted.bam
$samtools view tmp.rmdup.bam | cut -f 2 | sort | uniq -c
7 147
2 163
1 323
1 353
1 371
1 387
1 433
2 83
2 97
8 99
$samtools view tmp.sorted.bam | cut -f 2 | sort | uniq -c
8 147
3 163
1 323
1 353
1 371
1 387
1 433
3 83
2 97
9 99
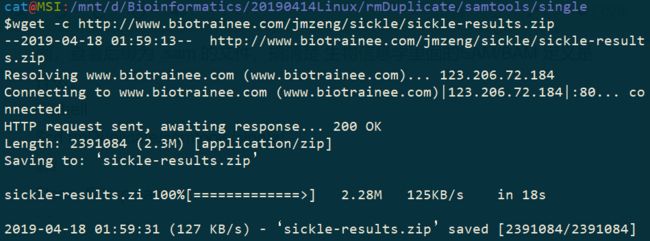
十六题、下载
http://www.biotrainee.com/jmzeng/sickle/sickle-results.zip文件,并且解压,查看里面的文件夹结构, 这个文件有2.3M,注意留心下载时间及下载速度。
$wget -c http://www.biotrainee.com/jmzeng/sickle/sickle-results.zip
$unzip sickle-results.zip
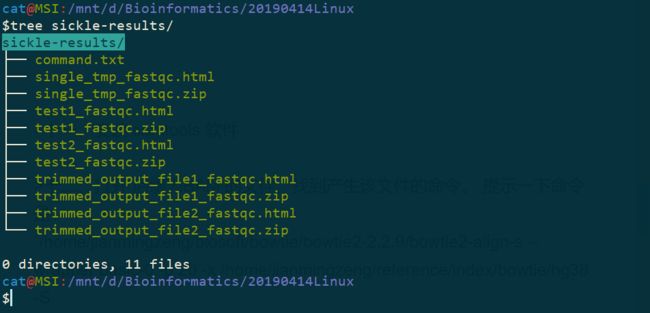
$tree sickle-results/
十七题、解压
sickle-results/single_tmp_fastqc.zip文件,并且进入解压后的文件夹,找到fastqc_data.txt文件,并且搜索该文本文件以>>开头的有多少行?
$cd sickle-results/
$ls
$unzip single_tmp_fastqc.zip
$cd single_tmp_fastqc/
$cat fastqc_data.txt | grep '>>' |wc -l
24
十八题、下载
http://www.biotrainee.com/jmzeng/tmp/hg38.tss文件,去NCBI找到TP53/BRCA1等自己感兴趣的基因对应的refseq数据库ID,然后找到它们的hg38.tss文件的哪一行。
https://www.ncbi.nlm.nih.gov/gene/7157
$wget -c http://www.biotrainee.com/jmzeng/tmp/hg38.tss
$less -SN hg38.tss

$cat hg38.tss | cut -f 1 | cut -d "_" -f 1 | sort | uniq -c
51064 NM
15954 NR
打开https://www.ncbi.nlm.nih.gov/gene/7157


总共有15条NM序列
NM_000546.5
NM_001126112.2
NM_001126113.2
NM_001126114.2
NM_001126115.1
NM_001126116.1
NM_001126117.1
NM_001126118.1
NM_001276695.1
NM_001276696.1
NM_001276697.1
NM_001276698.1
NM_001276699.1
NM_001276760.1
NM_001276761.1
$less -S hg38.tss | grep -n "NM_000546"
413:NM_000546 chr17 7685550 7689550 1
$grep -n "NM_000546" hg38.tss
413:NM_000546 chr17 7685550 7689550 1
十九题、解析
hg38.tss文件,统计每条染色体的基因个数。
$cat hg38.tss | cut -f 2 | cut -d "_" -f 1 | sort | uniq -c
6157 chr1
2838 chr10
3577 chr11
3014 chr12
1133 chr13
1982 chr14
2377 chr15
2696 chr16
3794 chr17
883 chr18
5880 chr19
4090 chr2
1692 chr20
895 chr21
1410 chr22
3395 chr3
2277 chr4
2821 chr5
5782 chr6
2785 chr7
2221 chr8
2310 chr9
2 chrM
32 chrUn
2561 chrX
414 chrY
二十题、解析
hg38.tss文件,统计NM和NR开头的熟练,了解NM和NR开头的含义。
$cat hg38.tss | cut -f 1 | cut -d "_" -f 1 | sort | uniq -c
51064 NM
15954 NR

参考资料:https://en.wikipedia.org/wiki/RefSeq