蛋白-小分子对接
1. 项目说明
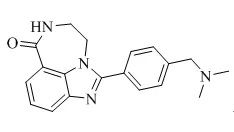
采用分子对接技术研究化合物1与受体PARP1的结合模式(图1)。
2.计算方法
从RCSB Protein Data Bank(http://www.rcsb.org)下载PARP1的X-ray晶体结构(PDB 编号:4RV6,分辨率:3.19 Å),以第一个构象作为受体结构。
[1].采用UCSF Chimera软件建立化合物1的三维结构,并进行能量优化。
[2,3].采用Dock Prep模块添加氢原子,并分别添加AMBER ff14SB力场和AM1-BCC电荷。采用Chimera中的DMS工具以半径为1.4 Å的探针生成受体的分子表面。
[4,5].X-ray晶体结构显示有1个合理的结合位点,对于该结合位点,使用sphgen模块生成围绕活性位点的球状集合(Spheres),使用Grid模块生成Grid文件,该文件用于基于Grid的能量打分评价。采用DOCK6.7程序进行半柔性对接(semi-flexible docking),生成10000个不同的构象取向(orientation)以及获得配体分子与结合位点的静电和范德华相互作用,并由此计算得到Grid打分。通过聚类分析(RMSD 阈值为2.0Å),得到打分最佳的构象。
[6].最后,采用PyMOL生成图片。
3.计算结果
A.结合构象打分
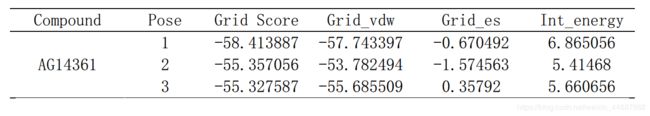
采用DOCK6.7程序预测化合物1在PARP1中的结合模式,保留最多20个结合构象。计算结果表明,结合位点均有多个对接构象,其打分情况如下(表1)。根据打分和结合模式选取第二个对接构象进行结合模式分析。
表1.化合物1与受体PARP1的对接打分(单位:kcal/mol)
B.结合模式分析
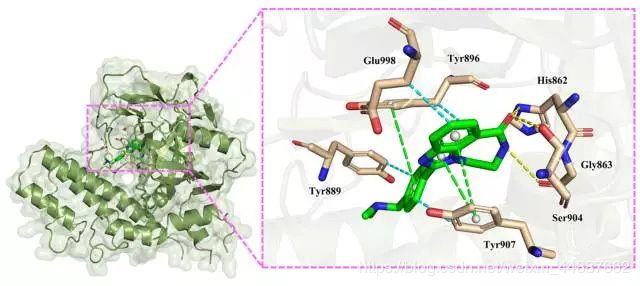
化合物1七元环上的酰胺羰基氧原子与氨基酸残基Ser904和His862形成氢键相互作用;同时,酰胺氮原子与Gly863形成了3.33 Å的氢键相互作用。这为化合物的结合锚定了方向并提供了一定的静电力贡献(Grid_es = -1.574563 kcal/mol)。
苯并咪唑的两个环与Tyr90之间形成P型π-π堆积作用,芳环中心距离分别为4.21 Å和4.93 Å;侧链苯环与Tyr896之间形成T型π-π堆积作用,芳环中心距离为5.47 Å。同时,化合物还与残基Tyr889、Tyr896、Tyr907和Glu998之间形成疏水作用,疏水作用和π-π堆积作用为化合物提供了强大的范德华力(Grid_vdw = -53.78 kcal/mol)。
综上,化合物1与蛋白PARP1的相互作用以π-π堆积和疏水作用为主,并通过氢键作用锁定结合取向。
参考文献:
[1].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE.Ucsf chimera–a visualization system for exploratory research and analysis.J Comput Chem,25(13):1605–12, 2004.
[2].Araz Jakalian,Bruce L.Bush,David B.Jack, and Christopher I.Bayly.Fast,efficient generationof high-quality atomic charges.am1-bcc model: I. method.Journal of Computational Chemistry,21(2):132–146, January 2000.
[3].Araz Jakalian, David B.Jack, and Christopher I. Bayly. Fast, efficient generation of high-quality atomic charges. am1-bcc model: I.parameterization and validation Journal of Computational
Chemistry,23(16):1623–1641, December 2002.
[4].P.Therese Lang,Scott R.Brozell,Sudipto Mukherjee,Eric F.Pettersen,Elaine C.Meng,Veena Thomas,Robert C.Rizzo,David A.Case, Thomas L.James James,and Irwin D.Kuntz.Dock 6:Combining techniques to model rna-small molecule complexes.RNA,5(6):1–12,December 2009.
[5].Sudipto Mukherjee, Trent E. Balius, and Robert C. Rizzo. Docking validation resources: Protein family and ligand flexibility experiments. Journal of Chemical Information and Modeling, 50(11):1986–2000, October 2010.
[6].Schrödinger,LLC.The PyMOL molecular graphics system, version 1.8,2015.
获取更多资讯,请关注以下微信公众号
