引言
以PacBio公司SMRT技术为代表的第三代单分子荧光测序平台,相较于传统NGS平台,虽测序费用较高,在CLR模式下测序错误率达5~15%,然而其长读长(十KB到几十KB),且在HiFi模式下测序准确,无需PCR扩增等优点为以测序为基础的科学研究带来了革命性的突破。
目前PB三代测序广泛应用于基因组Denovo、全长转录本检测、宏基因组、重测序和变异检测等多个方向,并且在染色体结构变异(SV)的检测中有着不可替代的优势。
今天起小编以信息分析的角度帮大家总结PB三代重测序基础和分析方法,期望各位看官读完之后能收获满满~
1.数据准备
1.1数据类型介绍
PB平台的subreads和CCS reads产生过程示意图如下:

由上述示意图可以看到,在PB三代测序产生数据的过程中,双链DNA片段两端添加测序接头,通过引物和DNA聚合酶的作用开始环形地边合成边测序过程,每合成一圈,可以得到2X DNA目的片段的测序结果。
测序结束后得到的包含若干圈含有接头和目的片段序列的reads,通常称之为polymerase reads,而去除测序接头后得到的若干条目的片段序列为subreads。
CLR模式:当插入片段读长较长时,一个插入片段只能测到一次,每一个ZMW(零模波导孔)得到的subreads就是该测序得到的序列,该序列的准确度在85%以上。
HiFi模式:当插入片段适中时,对于同一个片段会被测到N次,将来源于同一个ZMW(零模波导孔)的subreads 经过一致性矫正后会得到一条CCS read。由于CCS read经过一致性矫正,准确率非常高,在小突变的检测上也有极佳的表现。
1.2数据转化
CLR模式的下机数据,我们得到的是subreads。使用的下机文件是subreads.bam和subreads.bam.pbi,如下图所示:
subreads.bam:类似于NSG测序下机的fastq文件,以bam文件格式进行序列存储,减少占用储存空间和传输数据时间。
subreads.bam.pbi:是bam文件的索引文件,用于辅助比对分析更快地进行。
HiFi模式得到是更为准确的CCS reads,使用下机的ccs.bam和ccs.bam.pbi。如下图所示:- 小技巧:如果只有subreads.bam,怎么转换成ccs.bam呢?如果下机数据只有subreads.bam,并确认数据测序采用了HiFi模式,可使用PB官方smrtlink软件中的ccs工具将subreads.bam转换为ccs.bam,命令如下:
ccs *.subreads.bam *.ccs.bam
或者可以采用分割文件,并行模式,下面代码示例为分成10份运行:
ccs *.subreads.bam *.ccs.1.bam --chunk 1/10 -j
ccs *.subreads.bam *.ccs.2.bam --chunk 2/10 -j
...
ccs *.subreads.bam *.ccs.10.bam --chunk 10/10 -j
pbmerge -o *.ccs.bam movie.ccs.*.bam
转换过程非常慢,建议拆分并给足资源,千万别小气~
别忘了,转换完成后使用smrtlink中的pbindex工具生成.pbi索引文件:
pbindex *.ccs.bam
1.3参考基因组获取
分析前,除下机数据外,我们还需准备对应物种的参考基因组fasta文件。对此可以根据自己研究的需要,在NCBI、Ensembl、UCSC等常见数据库中进行下载,以UCSC下载人类hg19版本基因组序列为例:进入UCSC官网点击下载页签。
http://hgdownload.soe.ucsc.edu/downloads.html#human

点击上图红框所示,便可得到hg19的基因组序列。
2.数据比对

2.1比对方法介绍比较
在上述准备工作完成后,我们就可以将下机数据和我们下载的参考基因组序列进行比对了。
PB三代数据有很多比对软件可供选择,例如:LAST、BlasR、BWA-MEM、GraphMap、MECAT、minimap2、pbmm2、NGMLR等等,现在较为常用的是minimap2、pbmm2、NGMLR,其中pbmm2是PB官方基于minimap2进行优化的版本。
那么我们到底该选哪款?
别急,先让我们看看下面的文献测评结果:
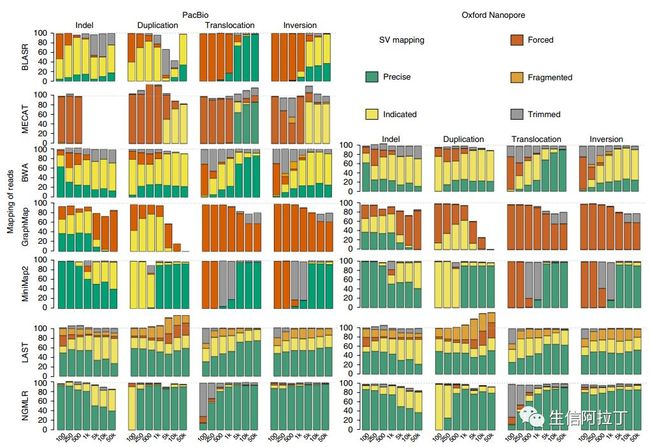
图中横坐标为不同结构变异的长度,由此我们可以看出,在进行结构变异检测时NGMLR比对效果要更好些,minimap2效果要稍逊。然而在实际项目分析时,NGMLR速度要慢于其他两款软件,且步骤稍繁琐,因此大家可以根据自己喜好,自行选择。
下面我们挑选两种常用软件,给大家展示一下~
2.2NGMLR比对分析
NGMLR比对命令:
如一个样品有多批数据需合并,需要如下操作,只有一批可跳过此步骤
samtools view -H *.bam -o header.sam
samtools merge -f -h header.sam out.bam input1.bam input2.bam
将bam文件转换为常规的fq文件,ngmlr不支持直接读入bam文件
bam2fastq --force out.bam -o out.fq
ngmlr比对并用samtools提取比对上reads进行输入,-t指定线程,-x 指定模式[pacbio,ont],-r 指定参考基因组(所在路径要有可写权限,否则无法正常生成.ngm文件,导致比对异常中断) -q 输入fq序列
ngmlr -t 20 -x pacbio -r ucsc.hg19.fasta -q out.fq 2 >> map.log | samtools view -h -bS -F 4 --o aligned.bam
使用samtools对比对后的bam文件进行排序,以便后续变异检测或其他处理
samtools sort -@ 4 -m 5G aligned.bam aligned.sort.bam
从上述分析步骤可以看出,由于ngmlr不支持多个输入文件,输出文件没有排序,没有选择性输出比对/未比对结果,因此整个步骤比较繁琐。如果整个过程使用4个线程进行分析,30X CLR数据预计耗时7天左右,20个线程约在1~2天内能完成分析。
2.3 PBMM2比对分析
pbmm2比对命令:
将同一个样品分批bam文件全路径逐行记录在fofn文件中
echo /your_path/1_subreads_or_ccs.bam,/your_path/2_subreads_or_ccs.bam,/your_path/3_subreads_or_ccs.bam | sed 's/,/\n/g' > test.bam.fofn
查看生成的文件,看看文件是否正确;此文件也可以手动创建
cat test.bam.fofn
/your_path/1_subreads_or_ccs.bam
/your_path/2_subreads_or_ccs.bam
/your_path/3_subreads_or_ccs.bam
运行pbmm2,可输入.fofn 或者单个bam文件
pbmm2 align ucsc.hg19.fa test.bam.fofn out.aligned.sort.bam(输出比对结果bam路径+名称) --sample aladdin -j 8 -J 8 --sort --preset CCS --median-filter --log-level INFO --log-file pbmm2.log
--sample指定样品名称,-j指定比对线程,-J 8指定排序步骤线程,--sort输出排序后的bam,--preset选择参数集,subreads数据请使用--preset SUBREADS
由于是PB官方推出的比对软件,整体和下机数据格式的契合度比较高,几乎可以实现一行命令从下机bam文件到比对后的bam文件,且无需再进行比对上的reads提取和排序。整个软件的速度也比较快,按照示例设置,1天内可以完成30X CLR数据分析。
小技巧
1.如果担心比对结果不佳,需要输出未比对的reads进行排查,可以加上--unmapped参数。
2.--median-filter需要注意,加入此参数,比对时默认来源于一个polymerase read(ZMW)的若干subreads,比对时只选用长度分布是这些reads长度中值的一条进行比对(猜测这可能是快的原因)。因此计算比对率时,公式应变更为:比对上的subreads数或ccs reads数/polymerase reads数,而不是其他软件惯用的除以输入的总reads数!
3.由于pbmm2生成的bam文件MD tag格式有些特别,无法用于后续的sniffles进行SV检测。实际测试使用samtools calmd 添加MD tag 可以用于sniffles分析。虽然可以运行,生成结果却异常,所以推荐使用pbsv进行后续SV检测。
关于minimap2的使用,限于篇幅,不再详述,相信大家看过前两个案例后,使用MinMap2不再话下。需要特别强调的是,如果使用minimap2 + sniffles进行SV检测,比对时一定要加上--MD来生成bam文件的MD tag,否则sniffles运行会报错!
今天,作为PB三代重测序系列的第一篇,从数据分析的角度介绍了三代重测序数据基本知识和比对分析。
写到此处不免生出“纸短情长”的感觉,关于PB三代重测序,还有太多的东西需要讲:染色体结构变异检测,小突变SNV和INDEL的检测、突变注释、多样品SV合并分析,肿瘤成对样品SV筛选、单基因病或遗传病家系分析等内容只能留待后续系列陆续推出,敬请期待!
最后啰嗦一句,喜欢的同学们不妨转发给更多需要的同胞,共同学习和进步是我们创刊更新的不竭动力,同时也欢迎大家留言讨论交流~
参考文献:
Sedlazeck F J , Rescheneder P , Smolka M , et al. Accurate detection of complex structural variations using single-molecule sequencing[J]. Nature Methods, 2018.
Audano P A, Sulovari A, Graves-Lindsay T A, et al. Characterizing the major structural variant alleles of the human genome[J]. Cell, 2019, 176(3): 663-675. e19.
Wenger A M, Peluso P, Rowell W J, et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome[J]. Nature biotechnology, 2019, 37(10): 1155-1162.
作者:云歌
审稿:童蒙
编辑:angelica

