2017/10/26 第一版:为什么要使用命令行,命令行和图形界面的差别。如何在命令行下处理数据的几个问题。
2017/10/26 第二版 : 增加几个题目的解释,AWK ,GREP ,SED和正则表达式开头。
2017/10/27 第三版:根据自己以前的笔记增加了Linux命令小结,用Linux命令处理数据,如何管理压缩文件。AWK部分语法
学习生信就不可避免要和命令行(Command line)打交道。大部分初学者“天生”排斥命令行,喜欢图形交互界面(GUI)。除了体型以外,选择软件真的也被基因所决定了吗?
图形软件 = 命令行 + 好看且直觉化的界面设计。
人类不是天生喜欢GUI,只是大脑天生喜欢偷懒,然后各种图形软件都是追求“一步到位”,你觉得自己只要拖拖拉拉点点就可以了。如果UI设计不太合理,甚至反人类,这个软件估计你用起来也感觉设计者是个弱智。还有很多图形软件的复杂度也是有区分的,比如说美图秀秀和图片商店。你会觉得自己用美图秀秀贼溜,但是图商里面连抠图都不会。其实从本质上看,美图秀秀只是将图商某些功能进行了整合,这些功能对于小白绝对够用,但是对于专业设计师而言,他们需要更加细节的把控。
在很多人眼里,生信无非就是跑流程。当然在做生信的人的眼里,湿实验无非就是跑PCR。那么问题来,博后和他带的师妹(弟)都跑PCR,区别在哪里呢?
无非就是明确自己究竟要解决什么问题,以及解决这个问题到底需要那几步。
Unix新兵训练营
请根据如下内容对Linux基础做一个回顾:
- The Terminal
- first unix command: ls
- The unix tree:
- finding where you are: pwd
- making new directories: mkdir
- Getting from 'A' to 'B': cd
- The root directory: /
- Navigating upwards in Unix filesystem: ..
- Absolute and relative paths
- Finding way back home: cd ~, cd
- making the ls commands more useful: ls -l, ls -R, ls -ltr, ls -lh
- man pages: man
- removing dirctories: rmdir , rm
- using TAB completion
- creating empty file with touch: touch
- moving files, mv
- renaming files, mv
- moving directories
- removing fiels, rm
- copying files, cp
- copying directories, cp
- viewing files with less or more
- viewing files with cat
- couting characters in a file, wc
- editng small text file with nano
- the $PATH environment variable
- matching lines in file with grep
- combining unix commands with pipe
Whenever making a long pipe, test each step as you build it!
- Miscellaneous Unix power commands
tail -n 20 file.txt | head
grep "^ATG" file.txt
cut -f 3 file.txt | sort -u
grep -c '[bc]at' file.txt
cat file.txt | tr 'a-z' 'A-Z'
cat file.txt | sed 's/Chr1/Chromosome 1/' > file2.txt
Unix重要概念: 流和重定向
Unix系统的设计理念其中之一就是:
This is the Unix philosophy: Write progams that do one thing and do it well. Write programs to work together. Wirte program to handle text streams, beacause that is a universal interface. --Doug McIlory
Unix的一个个命令就是流水线上的工人,只要做一件事情,并且把它做好。而按照目标把不同功能的工人进行整合就是使用管道符号|。比如说富土康流7号流水线的张全蛋负责对前面的结果进行质控。

每个工人在工作的时候如果遇到残次品(标准错误,stderr),可以重定向(>,>>,<,<<)到专门的地方进行存放。最终的结果可以是输出到屏幕,或者是重定向到文件中。
那么问题来了,Unix里有哪些管道命令。
注: 了解操作系统发展的人应该知道Linux是一种类Unix系统,没有Unix就没有后续BSD和Linux了。
管道命令

管道命令是一类能被管道符号|所连接的命令。它能够接受前一个命令标准输出作为自己的输入,处理后输出给下一个管道命令。常用的管道命令如下:
选取命令:
cut: cut 将一段信息的某一段切出来,处理的信息是以行为单位。
grep: grep 分析一行信息,如果有匹配的,就将该行拿出来。排序命令:
sort: sort 可以依据不同的数据类型进行排序。
uniq: uniq 重复的行只显示一个
wc: wc 输出信息的整体数据双向重定向
tee: tee 双重定向,存到文件/设备的同时,输出到屏幕以便继续处理。字符转换命令:
tr: tr 删除,压缩一段信息中的文字,或者进行文字信息的转换。
col: col 对特殊字符进行处理
join: join 将两个文件当中有相同数据的那一行加在一起。
paste: paste 将两个文件贴在一起,中间以[tab]键隔开。
expand: expand 将[tab]按键转成空格键切割命令:
split: 将大文件按照文件大小或者行数切割成小文件参数代换:
xargs: xargs 读入stdin的数据,并且以空格符或断行符进行分辨,将stdin的数据分隔为arguments。
不同的类Unix系统虽然可能都有这些命令,但是功能参数未必相同,比如说GNU就有一套。建议阅读GNU core utilitie 文档: https://www.gnu.org/software/coreutils/manual/coreutils.html
如何使用Unix命令处理数据
用head和tail,less查看数据
从第m行开始:tail -n +m file
到第m行结束: head -n +m file
同时看文件的开始和结尾: (head -n 2; tail -n 2 ) < file
BED 排序后,则可以看到位置特征
添加到配置文件(.bashrc)中
$ # inspect the first and last lines of a file
$ i() { (head -n 2; tail -n 2) < "$1" | column -t}
查找特定基因grep 'gene_id "xxxxx"' file.gt | head -n 1
利用head 结束后面不需要的搜索
less in more
wc,ls,awk对纯文本进行summary
查看行数(-l)和字符数(-m)
wc Mus_musculus.GRCm38.75_chr1.bed Mus_musculus.GRCm38.75_chr1.gtf
或
grep -c "[^ \\n\\t]" some_data.bed排除空行
文件大小: ls -lh file
查看列数:awk -F "\t" '{print NF; exit}' file 我们可以利用tail -n +x去掉前几行元信息数据,或grep -v "^#" file | head -n 1 | awk [...]
提取特定列: cut -f 2 file | head -n 3
cut, column格式化表格式数据
tab分隔的数据比较占空间,因此可以用column使其美观一点
$ grep -v "^#" file.gtf | cut -f 1,3,5 > file.bed
$ grep -v "^#" file.gtf | cut -f -8| column -t | head -n3
$ column -s ',' -t xxx.csv | head -n 3 #指定分隔符
全能的grep
pattern option:
-v : 反选
-w: 限制为word
-E:扩展正则表达式:grep -E "(Olfr1413|Olfr1411)" file
output option:
-n: 显示行
-B: 上文
-A: 下文
-C: 上下文
-c : 计算匹配数
-o : 只输出匹配部分,而非所在行
同一基因名因为包括转录本,外显子,起始密码子等,所以有许多冗余:
$ grep -e -o 'gene_id "(\w+)"' Mus_musculus.GRCm38.75_chr1.gtf | \
cut -f2 -d " " | \
sed 's/"//g"' | \
sort | \
uniq > mm_gene_id.txt
排序:sort
排序有利于提高效率,方便去重
sort默认使用空白作为间隔符,使用 -t 自定义间隔符
-k:排序关键字和顺序
-c: 检查是否排序过 sort -k1,1 -k2,2n -c example_sorted.bed
-r: 逆序
-S: 内存buffer -S2G
-V: 避免出现 chr1 chr22 chr3
--parallel: 使用核心 --parallel 4
# 两行逆序
sort -k1,1 -k,2n -r example.bed
#一行正序, 一行逆序
sort -k1,1 -k2,2nr example.bed
sort -k1,1V -k2,2n example.bed
去重 uniq
先排序后去重
uniq -c : 重复数
join
按照某一行组合不同文本,基本语法join -1
Unix’s join will not work unless both files are sorted by the column to join on.
sort -k1,1 example.txt > example_sorted.txt
sort -c -k1,1 example_length.txt > example_length_sorted.txt
join -1 1 -2 1 example_sorted.txt example_length_sorted.txt > example_with_length.txt
进阶shell技巧
subshells
subshells允许我们在一个独立的shell进程中执行sequential commands
echo "This command"; echo "that command" | sed 's/command/step/'
(echo "This command"; echo "that command") | sed 's/command/step'
管道命名和进程替换
# named pipe
mkfifo fqin
echo "hello named pipes" > fqin
cat fqin
## processes substition
program --in1 <(makein raw1.txt) --in2 <(makein raw2.txt)\
--out1 out1.txt --out2 out2.txt
program --in1 in1 --in2 in2 --out1 >(gzip >out1.txt.gz) --out2 >(gzip >out2.txt.gz)
管道命令处理数据Q&A
最重要的能力是提出问题的能力。
熟练掌握管道命令的独家秘诀就是看文档和做题。
每次开始项目时都应单独建立这些文件夹,避免混淆。
# 回到家目录
cd
mkdir edu src bin refs
数据来源: https://www.yeastgenome.org/,
链接分别为
- http://downloads.yeastgenome.org/curation/chromosomal_feature/SGD_features.tab
- http://downloads.yeastgenome.org/curation/chromosomal_feature/SGD_features.README
接下来使用Unix命令查看酵母基因组特征文件(genome feature files, gtf), gtf文件主要存放酵母基因组的注释信息。
如何下载数据?
wget http://downloads.yeastgenome.org/curation/chromosomal_feature/SGD_features.tab
# 数据说明
wget http://downloads.yeastgenome.org/curation/chromosomal_feature/SGD_features.README
如何了解数据的基本信息?
less SGD_features.README
# 逐页查看
less SGD_features.tab
如果我想看中间几行,比如说第50到60行,应该如何做? head只能看前几行,tail只能看几行,没有直接看中间几行的数据。但是问题可以分解成先看前60行,再看最后10行。
head -n 60 SGD_features.tab | tail -n 10
结果表明是酵母基因组注释信息。
有多少和感兴趣的目标匹配
grep YAL060W sc.tab | head
用颜色标明匹配
grep YAL060W sc.tab --color=always | head
# We can place the matching lines into a new file.
grep YAL060W sc.tab > match.tab
# The following is equivalent to the above.
# It is a bit longer and runs an extra program (cat).
# But it is cleaner and more clear what file gets opened (on the left).
# We use this construct for clarity even when shorter ones are available.
cat sc.tab | grep YAL060W > match.tab
# How many lines in the file match the word gene?
cat sc.tab | grep gene | wc -l
# The above matches the word gene anywhere on the line.
# It looks like this file uses the feature
# type (column 2) ORF for protein coding genes.
# Build your commands one step at a time.
# Pressing the up arrow recalls the previous line.
cat sc.tab | head
cat sc.tab | cut -f 2 | head
cat sc.tab | cut -f 2 | grep ORF | head
# We can get select multiple columns of interest from the output with cut.
cat sc.tab | cut -f 2,3,4 | grep ORF | head
# How many rows have the word ORF in the second column?
cat sc.tab | cut -f 2,3,4 | grep ORF | wc -l
剔除那些不匹配
cat sc.tab | grep -v Dubious | cut -f 2,5,9 | wc -l
有多少feature type
cat sc.tab | cut -f 2 > features.tab
## Sorting places identical consecutive entries next to one another.
cat features.tab | sort | head
## Find unique words. The uniq command collapses consecutive identical words into one.
cat features.tab | sort | uniq | head
##Using -c flag to uniq will not only collapse consecutive entires it will print their counts.
cat features.tab | sort | uniq -c | head
有多少个基因?有几种类型的基因注释信息?最先被注释的基因是哪一个,是什么时候?哪一种注释信息类型最多?
有多少个基因,也就是统计一共多少行(如果你担心重复,可以去重)
wc -l SGD_features.tab
# -l: 显示行数, 还有-m, 显示字符数, -b, 显示字节数
几种类型的基因注释信息: 先截取(cut)所在行,为了避免冗余先排序(sort)在去重(uniq),最后统计(wc)或者显示(nl)函数。
cat SGD_features.tab | cut -f 2 | sort | uniq | nl
最先被注释的基因是哪一个? 截取时间和基因名所在行,然后按照时间从小到大排序。
cut -f 1,14 SGD_features.tab | sort -k 2 | head
# -f, 表示选取第几列,比如说1-10,就是1到10列,1,3则是第一列和第三列
# -k, 表示用第几列作为排序对象
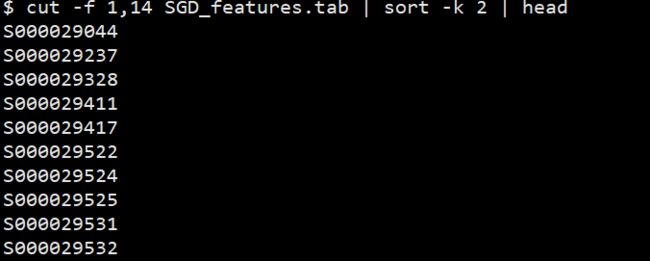
然后而是一片空白,说明有些基因是没有日期标注。因此需要用正则表达式(后续介绍)提取仅仅符合要求的列。
cut -f 1,14 SGD_features.tab | sort -k 2 | grep '.*[[:blank:]][[:alnum:]]' | head
# .*[[:blank:]][[:alnum:]], 表示任意多个字符后接着空白字符接着数字或者字符
[图片上传失败...(image-f604ef-1510145886606)]
如何处理压缩文件
压缩文件可以提高磁盘利用率,并且解压比压缩块不只一个数量级
压缩文件长什么样
一般如下结尾:.gz, .bz, .bz2, or .zip,或者是.tar.gz, .tar.bz2常见的压缩格式
ZIP: extension .zip, program names are zip/unzip. Available on most platforms.
GZIP: extension .gz, program names are gzip/gunzip. The most common compression format on Unix-like systems.
BZIP2: extension .bz/.bz2, program names are bzip2/bunzip2
XZ: extension .xz. A more recent invention. Technically bzip2 and (more so) xz are improvements over gzip, but gzip is still quite prevalent for historical reasons and because it requires less memory
生物信息学有没有特定的压缩格式
The
# Get a sequence file.
efetch -db=nuccore -format=fasta -id=AF086833 > AF086833.fa
bgzip AF086833.fa
如何压缩或解压文件呢
- 单个文件
# Get a sequence file.
efetch -db=nuccore -format=fasta -id=AF086833 > AF086833.fa
# Compress with gzip.
# Creates the file AF086833.fa.gz.
gzip AF086833.fa
# Some tools can operate on GZ files. If not you
# can also uncompress on "on the fly" without decompressing
# the entire file.
# Note: gzcat on macOS, zcat on Linux.
zcat AF086833.fa.gz | head
# Uncompress the file
# Creates the file AF086833.fa.
gunzip AF086833.fa.gz
- 多个文件
tar czfv sequences.tar.gz AF086833.fa AF086833.gb
tar czvf sequences.tar.gz AF086833.*
tar czvf sequences.tar.gz sequences/*
- 如何同步归档文件
tar -c sequences/* | gzip --rsyncable > file.tar.gz
What is a tarbomb?
tarbomb就是一个文档文件里面有许多文件,不建议直接解压到当前文件夹,应该专门建立一个文件夹
怎么再次使用tar?
我们会经常忘记应该用那些flag,所以多看手册吧。
