利用R语言对RNA-Seq进行探索分析与差异表达分析
最新更新见 http://cangfengzhe.github.io/r/RNA-Seq.html
介绍
本文参考 bioconductor 中RNA-Seq workflow: gene-level exploratory analysis and differential expression并对其根据需要进行了增减。
更多细节还请参考 http://www.bioconductor.org/help/workflows/rnaseqGene/
试验数据
数据来源
Himes BE, Jiang X, Wagner P, Hu R, Wang Q, Klanderman B, Whitaker RM, Duan Q, Lasky-Su J, Nikolos C, Jester W, Johnson M, Panettieri R Jr, Tantisira KG, Weiss ST, Lu Q. “RNA-Seq Transcriptome Profiling Identifies CRISPLD2 as a Glucocorticoid Responsive Gene that Modulates Cytokine Function in Airway Smooth Muscle Cells.” PLoS One. 2014 Jun 13;9(6):e99625. PMID: 24926665. GEO: GSE52778.
在这个RNA-Seq试验中,采用了4种呼吸道平滑肌肉细胞(airway smooth muscle cells),每种细胞均有 地塞米松治疗、非治疗两类。共计8个样本,储存在 airway 包中。
原始数据的处理
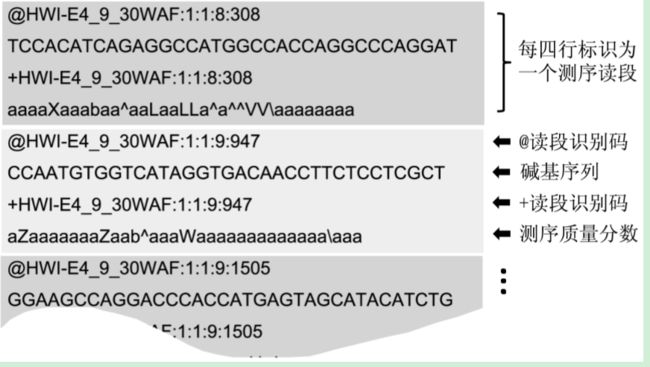
高通量测序数据常采用 FASTQ 格式来保 存所测的碱基读段和质量分数。如图 所示,FASTQ 格式以测序读段为单位存 储,每条读段占 4 行,其中第一行和的第三行由文件识别标志和读段名(ID)组成(第一行以“@”开头而第三行以“+”开头;第三行中 ID 可以省略,但“+”不能省 略),第二行为碱基序列,第四行为各碱基所对应的测序质量分数序列。
采用 tophat/bowtie2 将原始数据fastq映射到基因组序列,得到bam文件;
此处我们采用airway 自带的bam文件。
加载 airway 包, 并利用自带bam文件
library("airway")
dir <- system.file("extdata", package="airway", mustWork=TRUE)
list.files(dir) # 文件列表## [1] "GSE52778_series_matrix.txt" "Homo_sapiens.GRCh37.75_subset.gtf"
## [3] "sample_table.csv" "SraRunInfo_SRP033351.csv"
## [5] "SRR1039508_subset.bam" "SRR1039508_subset.bam.bai"
## [7] "SRR1039509_subset.bam" "SRR1039512_subset.bam"
## [9] "SRR1039513_subset.bam" "SRR1039516_subset.bam"
## [11] "SRR1039517_subset.bam" "SRR1039520_subset.bam"
## [13] "SRR1039521_subset.bam"csvfile <- file.path(dir,"sample_table.csv")
(sampleTable <- read.csv(csvfile,row.names=1)) # 获取样本信息## SampleName cell dex albut Run avgLength Experiment Sample BioSample
## SRR1039508 GSM1275862 N61311 untrt untrt SRR1039508 126 SRX384345 SRS508568 SAMN02422669
## SRR1039509 GSM1275863 N61311 trt untrt SRR1039509 126 SRX384346 SRS508567 SAMN02422675
## SRR1039512 GSM1275866 N052611 untrt untrt SRR1039512 126 SRX384349 SRS508571 SAMN02422678
## SRR1039513 GSM1275867 N052611 trt untrt SRR1039513 87 SRX384350 SRS508572 SAMN02422670
## SRR1039516 GSM1275870 N080611 untrt untrt SRR1039516 120 SRX384353 SRS508575 SAMN02422682
## SRR1039517 GSM1275871 N080611 trt untrt SRR1039517 126 SRX384354 SRS508576 SAMN02422673
## SRR1039520 GSM1275874 N061011 untrt untrt SRR1039520 101 SRX384357 SRS508579 SAMN02422683
## SRR1039521 GSM1275875 N061011 trt untrt SRR1039521 98 SRX384358 SRS508580 SAMN02422677filenames <- file.path(dir, paste0(sampleTable$Run, "_subset.bam")) # 提取bam文件获取bam数据
library("Rsamtools")
filenames ## [1] "/Library/Frameworks/R.framework/Versions/library/airway/extdata/SRR1039508_subset.bam"
## [2] "/Library/Frameworks/R.framework/Versions/library/airway/extdata/SRR1039509_subset.bam"
## [3] "/Library/Frameworks/R.framework/Versions/library/airway/extdata/SRR1039512_subset.bam"
## [4] "/Library/Frameworks/R.framework/Versions/library/airway/extdata/SRR1039513_subset.bam"
## [5] "/Library/Frameworks/R.framework/Versions/library/airway/extdata/SRR1039516_subset.bam"
## [6] "/Library/Frameworks/R.framework/Versions/library/airway/extdata/SRR1039517_subset.bam"
## [7] "/Library/Frameworks/R.framework/Versions/library/airway/extdata/SRR1039520_subset.bam"
## [8] "/Library/Frameworks/R.framework/Versions/library/airway/extdata/SRR1039521_subset.bam"bamfiles <- BamFileList(filenames, yieldSize=2000000) # 将bam文件放入列表,yieldSize 表示每次被读取的记录数
seqinfo(bamfiles) # 序列的基本信息## Seqinfo object with 84 sequences from an unspecified genome:
## seqnames seqlengths isCircular genome
## 1 249250621
## 10 135534747
## 11 135006516
## 12 133851895
## 13 115169878
## ... ... ... ...
## GL000210.1 27682
## GL000231.1 27386
## GL000229.1 19913
## GL000226.1 15008
## GL000207.1 4262 seqlevels(bamfiles) # 所有的染色体名称, GLxxxxxx.x表示genomic contig## [1] "1" "10" "11" "12" "13" "14" "15"
## [8] "16" "17" "18" "19" "2" "20" "21"
## [15] "22" "3" "4" "5" "6" "7" "8"
## [22] "9" "MT" "X" "Y" "GL000192.1" "GL000225.1" "GL000194.1"
## [29] "GL000193.1" "GL000200.1" "GL000222.1" "GL000212.1" "GL000195.1" "GL000223.1" "GL000224.1"
## [36] "GL000219.1" "GL000205.1" "GL000215.1" "GL000216.1" "GL000217.1" "GL000199.1" "GL000211.1"
## [43] "GL000213.1" "GL000220.1" "GL000218.1" "GL000209.1" "GL000221.1" "GL000214.1" "GL000228.1"
## [50] "GL000227.1" "GL000191.1" "GL000208.1" "GL000198.1" "GL000204.1" "GL000233.1" "GL000237.1"
## [57] "GL000230.1" "GL000242.1" "GL000243.1" "GL000241.1" "GL000236.1" "GL000240.1" "GL000206.1"
## [64] "GL000232.1" "GL000234.1" "GL000202.1" "GL000238.1" "GL000244.1" "GL000248.1" "GL000196.1"
## [71] "GL000249.1" "GL000246.1" "GL000203.1" "GL000197.1" "GL000245.1" "GL000247.1" "GL000201.1"
## [78] "GL000235.1" "GL000239.1" "GL000210.1" "GL000231.1" "GL000229.1" "GL000226.1" "GL000207.1"导入基因组特征(注释)
eg. 外显子的染色体位置, 基因的起始、终止位点
library("GenomicFeatures")
gtffile <- file.path(dir,"Homo_sapiens.GRCh37.75_subset.gtf")
(txdb <- makeTxDbFromGFF(gtffile, format="gtf"))## TxDb object:
## # Db type: TxDb
## # Supporting package: GenomicFeatures
## # Data source: /Library/Frameworks/R.framework/Versions/library/airway/extdata/Homo_sapiens.GRCh37.75_subset.gtf
## # Organism: NA
## # miRBase build ID: NA
## # Genome: NA
## # transcript_nrow: 65
## # exon_nrow: 279
## # cds_nrow: 158
## # Db created by: GenomicFeatures package from Bioconductor
## # Creation time: 2015-06-17 17:40:06 +0800 (Wed, 17 Jun 2015)
## # GenomicFeatures version at creation time: 1.20.1
## # RSQLite version at creation time: 1.0.0
## # DBSCHEMAVERSION: 1.1(genes <- exonsBy(txdb, by="gene"))## GRangesList object of length 20:
## $ENSG00000009724
## GRanges object with 18 ranges and 2 metadata columns:
## seqnames ranges strand | exon_id exon_name
## |
## [1] 1 [11086580, 11087705] - | 98 ENSE00000818830
## [2] 1 [11090233, 11090307] - | 99 ENSE00000472123
## [3] 1 [11090805, 11090939] - | 100 ENSE00000743084
## [4] 1 [11094885, 11094963] - | 101 ENSE00000743085
## [5] 1 [11097750, 11097868] - | 103 ENSE00003520086
## ... ... ... ... ... ... ...
## [14] 1 [11106948, 11107176] - | 111 ENSE00003467404
## [15] 1 [11106948, 11107176] - | 112 ENSE00003489217
## [16] 1 [11107260, 11107280] - | 113 ENSE00001833377
## [17] 1 [11107260, 11107284] - | 114 ENSE00001472289
## [18] 1 [11107260, 11107290] - | 115 ENSE00001881401
##
## ...
## <19 more elements>
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengthsseqlevels(genes) # 染色体的名字## [1] "1"染色体的名称 seqlevels(bamfiles) 与 seqlevels(genes) 应该保持一致, 特别留意是否含有 “chr”, 要么都有”chr”, 要么都没有。
注意,这里采用了一个基因组注释文件的子集, 完整的信息可以从
ftp://ftp.ensembl.org/pub/release-75/gtf/ 获取
基因计数
library("GenomicAlignments")
se <- summarizeOverlaps(features = genes, reads = bamfiles,
mode = "Union", # 读段覆盖的模式
singleEnd=FALSE, #双末端 not 单末端
ignore.strand=TRUE,# True 表示忽略±链的限制
fragments=TRUE ) # 只应用于双末端测序,true表示非成对的对端应该被计数
class(se) # 得到 SummarizedExperiment 数据,可用于后续计算## [1] "SummarizedExperiment"
## attr(,"package")
## [1] "GenomicRanges"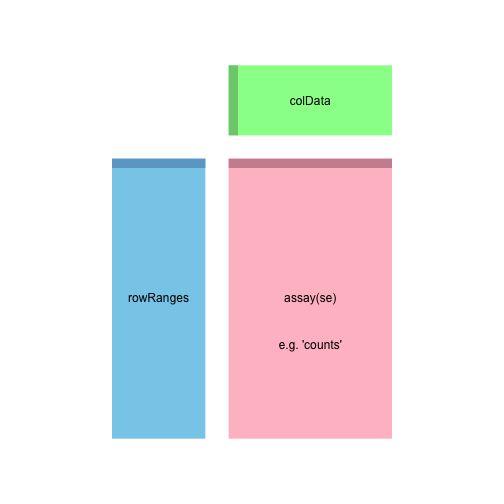
上图显示的是SummarizedExperiment类(以及他的子类DESeqDataSet)的布局, 粉红色 assay(se) 表示实际的数据, 每行为一个基因,每列为一个样本;
colData 表示样本的具体信息,随后我们会对它进行填充;rowRanges 表示每一个基因的信息。具体如下
se## class: SummarizedExperiment
## dim: 20 8
## exptData(0):
## assays(1): counts
## rownames(20): ENSG00000009724 ENSG00000116649 ... ENSG00000271794 ENSG00000271895
## rowRanges metadata column names(0):
## colnames(8): SRR1039508_subset.bam SRR1039509_subset.bam ... SRR1039520_subset.bam
## SRR1039521_subset.bam
## colData names(0):head(assay(se))## SRR1039508_subset.bam SRR1039509_subset.bam SRR1039512_subset.bam
## ENSG00000009724 38 28 66
## ENSG00000116649 1004 1255 1122
## ENSG00000120942 218 256 233
## ENSG00000120948 2751 2080 3353
## ENSG00000171819 4 50 19
## ENSG00000171824 869 1075 1115
## SRR1039513_subset.bam SRR1039516_subset.bam SRR1039517_subset.bam
## ENSG00000009724 24 42 41
## ENSG00000116649 1313 1100 1879
## ENSG00000120942 252 269 465
## ENSG00000120948 1614 3519 3716
## ENSG00000171819 543 1 10
## ENSG00000171824 1051 944 1405
## SRR1039520_subset.bam SRR1039521_subset.bam
## ENSG00000009724 47 36
## ENSG00000116649 745 1536
## ENSG00000120942 207 400
## ENSG00000120948 2220 1990
## ENSG00000171819 14 1067
## ENSG00000171824 748 1590colSums(assay(se))## SRR1039508_subset.bam SRR1039509_subset.bam SRR1039512_subset.bam SRR1039513_subset.bam
## 6478 6501 7699 6801
## SRR1039516_subset.bam SRR1039517_subset.bam SRR1039520_subset.bam SRR1039521_subset.bam
## 8009 10849 5254 9168colData(se)## DataFrame with 8 rows and 0 columnsrowRanges(se)## GRangesList object of length 20:
## $ENSG00000009724
## GRanges object with 18 ranges and 2 metadata columns:
## seqnames ranges strand | exon_id exon_name
## |
## [1] 1 [11086580, 11087705] - | 98 ENSE00000818830
## [2] 1 [11090233, 11090307] - | 99 ENSE00000472123
## [3] 1 [11090805, 11090939] - | 100 ENSE00000743084
## [4] 1 [11094885, 11094963] - | 101 ENSE00000743085
## [5] 1 [11097750, 11097868] - | 103 ENSE00003520086
## ... ... ... ... ... ... ...
## [14] 1 [11106948, 11107176] - | 111 ENSE00003467404
## [15] 1 [11106948, 11107176] - | 112 ENSE00003489217
## [16] 1 [11107260, 11107280] - | 113 ENSE00001833377
## [17] 1 [11107260, 11107284] - | 114 ENSE00001472289
## [18] 1 [11107260, 11107290] - | 115 ENSE00001881401
##
## ...
## <19 more elements>
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengthsstr(metadata(rowRanges(se)))## List of 1
## $ genomeInfo:List of 14
## ..$ Db type : chr "TxDb"
## ..$ Supporting package : chr "GenomicFeatures"
## ..$ Data source : chr "/Library/Frameworks/R.framework/Versions/library/airway/extdata/Homo_sapiens.GRCh37.75_subset.gtf"
## ..$ Organism : chr NA
## ..$ miRBase build ID : chr NA
## ..$ Genome : chr NA
## ..$ transcript_nrow : chr "65"
## ..$ exon_nrow : chr "279"
## ..$ cds_nrow : chr "158"
## ..$ Db created by : chr "GenomicFeatures package from Bioconductor"
## ..$ Creation time : chr "2015-06-17 17:40:06 +0800 (Wed, 17 Jun 2015)"
## ..$ GenomicFeatures version at creation time: chr "1.20.1"
## ..$ RSQLite version at creation time : chr "1.0.0"
## ..$ DBSCHEMAVERSION : chr "1.1"(colData(se) <- DataFrame(sampleTable)) # 填充样本的具体信息,方便后续分组,寻找差异基因## DataFrame with 8 rows and 9 columns
## SampleName cell dex albut Run avgLength Experiment Sample
##
## SRR1039508 GSM1275862 N61311 untrt untrt SRR1039508 126 SRX384345 SRS508568
## SRR1039509 GSM1275863 N61311 trt untrt SRR1039509 126 SRX384346 SRS508567
## SRR1039512 GSM1275866 N052611 untrt untrt SRR1039512 126 SRX384349 SRS508571
## SRR1039513 GSM1275867 N052611 trt untrt SRR1039513 87 SRX384350 SRS508572
## SRR1039516 GSM1275870 N080611 untrt untrt SRR1039516 120 SRX384353 SRS508575
## SRR1039517 GSM1275871 N080611 trt untrt SRR1039517 126 SRX384354 SRS508576
## SRR1039520 GSM1275874 N061011 untrt untrt SRR1039520 101 SRX384357 SRS508579
## SRR1039521 GSM1275875 N061011 trt untrt SRR1039521 98 SRX384358 SRS508580
## BioSample
## 注意:此处得到的数据需要采用EDSeq2包进行差异分析,所以不对数据进行标准化,切记。
差异表达基因分析
我们采用 DESeq2 包进行,差异表达基因的分析
# 此步采用 airway 包自带的se数据进行后续操作,可以忽略。如果没有进行上面的步骤也可以直接采用下面的数据进行后续操作。
data("airway")
se <- airway
library("DESeq2")
dds <- DESeqDataSet(se, design = ~ cell + dex) # design 参数为 formula,此处为cell和dex两个因素,~ cell + dex表示我们想控制cell研究dex的影响。采用DESeqDataSetFromMatrix函数从matrix中获取数据
countdata <- assay(se) # 可以根据自己的需要填充自己的数据(matrix格式),这里以assay(se)为例
class(countdata)
head(countdata)
coldata <- colData(se)
(ddsMat <- DESeqDataSetFromMatrix(countData = countdata,
colData = coldata,
design = ~ cell + dex))
dds$dex <- relevel(dds$dex, "untrt") # 将 untrt 定义为dex因素的第一水平,随后的foldchange 将采用 trt/untrt
dds <- DESeq(dds)
(res <- results(dds)) # 得到结果,可以根据padj来挑选合适的差异表达基因,log2FoldChange来确定基因上调还是下调,pvalue的校正采用了Benjamini-Hochberg方法,具体见 ?p.adjust## log2 fold change (MAP): dex trt vs untrt
## Wald test p-value: dex trt vs untrt
## DataFrame with 20 rows and 6 columns
## baseMean log2FoldChange lfcSE stat pvalue padj
##
## ENSG00000009724 41.75438 -0.9605961 0.25246721 -3.804835 1.418988e-04 3.783967e-04
## ENSG00000116649 1222.28667 0.2071285 0.07112287 2.912263 3.588209e-03 7.176417e-03
## ENSG00000120942 280.89869 0.1403078 0.10305342 1.361505 1.733540e-01 2.311387e-01
## ENSG00000120948 2698.77923 -0.7665547 0.14936634 -5.132044 2.866120e-07 1.146448e-06
## ENSG00000171819 188.08482 4.3627872 0.57110974 7.639140 2.186777e-14 1.749421e-13
## ... ... ... ... ... ... ...
## ENSG00000238199 0.3913511 -0.3733789 1.6308428 -0.2289484 0.81890903 NA
## ENSG00000253086 0.1518861 -1.0350645 1.5729023 -0.6580603 0.51049939 NA
## ENSG00000264181 0.0000000 NA NA NA NA NA
## ENSG00000271794 0.0000000 NA NA NA NA NA
## ENSG00000271895 34.9593217 -0.5451150 0.2920692 -1.8663899 0.06198683 0.09917893mcols(res, use.names=TRUE)## DataFrame with 6 rows and 2 columns
## type description
##
## baseMean intermediate mean of normalized counts for all samples
## log2FoldChange results log2 fold change (MAP): dex trt vs untrt
## lfcSE results standard error: dex trt vs untrt
## stat results Wald statistic: dex trt vs untrt
## pvalue results Wald test p-value: dex trt vs untrt
## padj results BH adjusted p-valuessummary(res)##
## out of 16 with nonzero total read count
## adjusted p-value < 0.1
## LFC > 0 (up) : 2, 12%
## LFC < 0 (down) : 3, 19%
## outliers [1] : 0, 0%
## low counts [2] : 8, 50%
## (mean count < 12.1)
## [1] see 'cooksCutoff' argument of ?results
## [2] see 'independentFiltering' argument of ?results保存数据
write.csv(res, file = '/your/path/')sessionInfo
sessionInfo()## R version 3.2.0 (2015-04-16)
## Platform: x86_64-apple-darwin13.4.0 (64-bit)
## Running under: OS X 10.10.3 (Yosemite)
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] parallel stats4 graphics grDevices utils datasets stats methods base
##
## other attached packages:
## [1] GenomicAlignments_1.4.1 GenomicFeatures_1.20.1 AnnotationDbi_1.30.1
## [4] Biobase_2.28.0 knitr_1.10.5 BiocStyle_1.6.0
## [7] Rsamtools_1.20.4 Biostrings_2.36.1 XVector_0.8.0
## [10] airway_0.102.0 DESeq2_1.8.1 RcppArmadillo_0.5.200.1.0
## [13] Rcpp_0.11.6 GenomicRanges_1.20.5 GenomeInfoDb_1.4.0
## [16] IRanges_2.2.4 S4Vectors_0.6.0 BiocGenerics_0.14.0
## [19] readr_0.1.1 sqldf_0.4-10 RSQLite_1.0.0
## [22] DBI_0.3.1 gsubfn_0.6-6 proto_0.3-10
## [25] dplyr_0.4.1 plyr_1.8.3
##
## loaded via a namespace (and not attached):
## [1] splines_3.2.0 Formula_1.2-1 assertthat_0.1 latticeExtra_0.6-26
## [5] yaml_2.1.13 lattice_0.20-31 chron_2.3-45 digest_0.6.8
## [9] RColorBrewer_1.1-2 colorspace_1.2-6 htmltools_0.2.6 XML_3.98-1.2
## [13] biomaRt_2.24.0 genefilter_1.50.0 zlibbioc_1.14.0 xtable_1.7-4
## [17] snow_0.3-13 scales_0.2.5 BiocParallel_1.2.3 annotate_1.46.0
## [21] ggplot2_1.0.1 nnet_7.3-9 survival_2.38-1 magrittr_1.5
## [25] evaluate_0.7 MASS_7.3-40 foreign_0.8-63 tools_3.2.0
## [29] formatR_1.2 stringr_1.0.0.9000 munsell_0.4.2 locfit_1.5-9.1
## [33] cluster_2.0.1 lambda.r_1.1.7 futile.logger_1.4.1 grid_3.2.0
## [37] RCurl_1.95-4.6 bitops_1.0-6 tcltk_3.2.0 rmarkdown_0.7
## [41] gtable_0.1.2 reshape2_1.4.1 gridExtra_0.9.1 rtracklayer_1.28.4
## [45] Hmisc_3.16-0 futile.options_1.0.0 stringi_0.4-1 geneplotter_1.46.0
## [49] rpart_4.1-9 acepack_1.3-3.3library(knitr)
knit('/Users/lipidong/baiduyun/work/RFile/MarkDown/funSet.Rmd', output = '~/learn/blog/_posts/2015-06-17-RNA-Seq.md')