生信小白学习日记Day7——WGS分析流程(picard)
2019年6月2日,周日,天气晴,pass 上午。开始学习NGS分析,继BWA比对和SAM文件排序转BAM后的流程。
NGS分析
step5 Mark Duplications
参考这篇:GATK使用方法详解。https://www.plob.org/article/7009.html
在制备文库的过程中,由于PCR扩增过程中会存在一些偏差,也就是说有的序列会被过量扩增。这样,在比对的时候,这些过量扩增出来的完全相同的序列就会比对到基因组的相同位置。而这些过量扩增的reads并不是基因组自身固有序列,不能作为变异检测的证据,因此,要尽量去除这些由PCR扩增所形成的duplicates,这一步可以使用picard-tools来完成。去重复的过程是给这些序列设置一个flag以标志它们,方便GATK的识别。还可以设置 REMOVE_DUPLICATES=true 来丢弃duplicated序列。对于是否选择标记或者删除,对结果应该没有什么影响,GATK官方流程里面给出的例子是仅做标记不删除。这里定义的重复序列是这样的:如果两条reads具有相同的长度而且比对到了基因组的同一位置,那么就认为这样的reads是由PCR扩增而来,就会被GATK标记。
e.g.:
java -jar picard-tools-1.96/MarkDuplicates.jar
REMOVE_DUPLICATES= false
MAX_FILE_HANDLES_FOR_READ_ENDS_MAP=8000
INPUT=hg19.reorder.sort.addhead_03.bam
OUTPUT=hg19.reorder.sort.addhead.dedup_04.bam METRICS_FILE=hg19.reorder.sort.addhead.dedup_04.metrics
实验室常用脚本中还会加入:VALIDATION_STRINGENCY=LENIENT
去查了一下,不知道理解的对不对:在BWA 比对生成SAM文件时,将没有map到基因组上的read归到了ref以外的区域,其MAPQ值不为0,而Picard认为这些read是不应该出现的,所以会报错(“MAPQ should be 0 for unmapped read” or “CIGAR should have zero elements for unmapped read”)。如果想忽略报错的话,就使用这行代码。
BWA can produce SAM records that are marked as unmapped but have non-zero MAPQ and/or non-"*" CIGAR. Typically this is because BWA found an alignment for the read that hangs off the end of the reference sequence. Picard considers such input to be invalid. In general, this error can be suppressed in Picard programs by passing VALIDATION_STRINGENCY=LENIENT or VALIDATION_STRINGENCY=SILENT. For ValidateSamFile, you can pass the arguments IGNORE=INVALID_MAPPING_QUALITY IGNORE=INVALID_CIGAR.(https://sourceforge.net/p/picard/wiki/Main_Page/)
注意: dedup这一步只要在library层面上进行就可以了,例如一个sample如果建了多个库的话,对每个库进行dedup即可,不需要把所有库合成一个sample再进行dedup操作。其实并不能准确的定义被mask的reads到底是不是duplicates,重复序列的程度与测序深度和文库类型都有关系。最主要目的就是尽量减小文库构建时引入文库的PCR bias。
Step 6: Index Bam File
对上一步得到的结果生成索引文件, 可以用samtools完成,生成的索引后缀是bai。
e.g.
samtools index hg19.reorder.sort.addhead.dedup_04.bam
Step 7: Local realignment around indels
这一步的目的就是将比对到indel附近的reads进行局部重新比对,将比对的错误率降到最低。一般来说,绝大部分需要进行重新比对的基因组区域,都是因为插入/缺失的存在,因为在indel附近的比对会出现大量的碱基错配,这些碱基的错配很容易被误认为SNP。还有,在比对过程中,比对算法对于每一条read的处理都是独立的,不可能同时把多条reads与参考基因组比对来排错。因此,即使有一些reads能够正确的比对到indel,但那些恰恰比对到indel开始或者结束位置的read也会有很高的比对错误率,这都是需要重新比对的。Local realignment就是将由indel导致错配的区域进行重新比对,将indel附近的比对错误率降到最低。
主要分为两步:
第一步,通过运行RealignerTargetCreator来确定要进行重新比对的区域。
e.g.
java -jar GenomeAnalysisTK.jar
-R hg19.fa
-T RealignerTargetCreator
-I hg19.reorder.sort.addhead.dedup_04.bam
-o hg19.dedup.realn_06.intervals
-known Mills_and_1000G_gold_standard.indels.hg19.vcf
-known 1000G_phase1.indels.hg19.vcf
在实验室常用脚本中,-T RealignerTargetCreator 会加入参数 -nt 2 没查到有什么作用;且没有后面的两个-known。
参数说明:
-R: 参考基因组;
-T: 选择的GATK工具;
-I: 输入上一步所得bam文件;
-o: 输出的需要重新比对的基因组区域结果;
-maxInterval: 允许进行重新比对的基因组区域的最大值,不能太大,太大耗费会很长时间,默认500;
-known: 已知的可靠的indel位点,重比对将主要围绕这些位点进行,对于人类基因组数据而言,可以直接指定GATK resource bundle里面的indel文件(必须是vcf文件)。
对于known sites的选择很重要,GATK中每一个用到known sites的工具对于known sites的使用都是不一样的,但是所有的都有一个共同目的,那就是分辨真实的变异位点和不可信的变异位点。如果不提供这些known sites的话,这些统计工具就会产生偏差,最后会严重影响结果的可信度。在这些需要知道known sites的工具里面,只有UnifiedGenotyper和HaplotypeCaller对known sites没有太严格的要求。
如果你所研究的对象是人类基因组的话,那就简单多了,因为GATK网站上对如何使用人类基因组的known sites做出了详细的说明,具体的选择方法如下表,这些文件都可以在GATK resource bundle中下载。
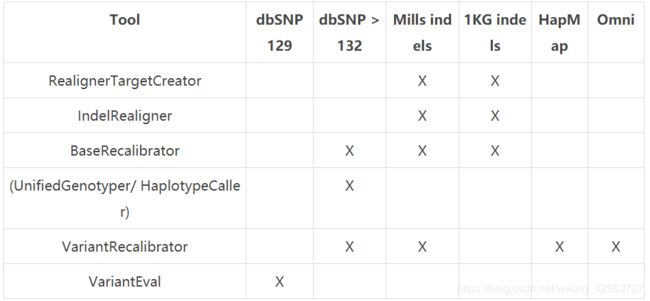
但是如果你要研究的不是人类基因组的话,那就有点麻烦了,http://www.broadinstitute.org/gatk/guide/article?id=1243,这个网站上是做非人类基因组时,大家分享的经验,可以参考一下。这个known sites如果实在没有的话,也是可以自己构建的:首先,先使用没有经过矫正的数据进行一轮SNP calling;然后,挑选最可信的SNP位点进行BQSR分析;最后,在使用这些经过BQSR的数据进行一次真正的SNP calling。这几步可能要重复好多次才能得到可靠的结果。
第二步,通过运行IndelRealigner在这些区域内进行重新比对。
e.g.
java -jar GenomeAnalysisTK.jar
-R hg19.fa
-T IndelRealigner
-targetIntervals hg19.dedup.realn_06.intervals
-I hg19.reorder.sort.addhead.dedup_04.bam
-o hg19.dedup.realn_07.bam
-known Mills_and_1000G_gold_standard.indels.hg19.vcf
-known 1000G_phase1.indels.hg19.vcf
运行结束后,生成的hg19.dedup.realn_07.bam即为最后重比对后的文件。
注意:
-
第一步和第二步中使用的输入文件(bam文件)、参考基因组和已知indel文件必须是相同的文件。
-
当在相同的基因组区域发现多个indel存在时,这个工具会从其中选择一个最有可能存在比对错误的indel进行重新比对,剩余的其他indel不予考虑。
-
对于454下机数据,本工具不支持。此外,这一步还会忽略bwa比对中质量值为0的read以及在CIGAR信息中存在连续indel的reads。
Step 8: Base quality score recalibration
注:实验室脚本中是没有这一步的,原因可能是我们没有known的.vcf文件。
这一步是对bam文件里reads的碱基质量值进行重新校正,使最后输出的bam文件中reads中碱基的质量值能够更加接近真实的与参考基因组之间错配的概率。这一步适用于多种数据类型,包括illunima、solid、454、CG等数据格式。在GATK2.0以上版本中还可以对indel的质量值进行校正,这一步对indel calling非常有帮助
举例说明,在reads碱基质量值被校正之前,我们要保留质量值在Q25以上的碱基,但是实际上质量值在Q25的这些碱基的错误率在1%,也就是说质量值只有Q20,这样就会对后续的变异检测的可信度造成影响。还有,在边合成边测序的测序过程中,在reads末端碱基的错误率往往要比起始部位更高。另外,AC的质量值往往要低于TG。BQSR的就是要对这些质量值进行校正。
BQSR主要有三步:
第一步:利用工具BaseRecalibrator,根据一些known sites,生成一个校正质量值所需要的数据文件,GATK网站以“.grp”为后缀命名。
e.g.
java -jar GenomeAnalysisTK.jar
-T BaseRecalibrator
-R hg19.fa
-I ChrALL.100.sam.dedup.realn.07.bam
-knownSites dbsnp_137.hg19.vcf
-knownSites Mills_and_1000G_gold_standard.indels.hg19.vcf
-knownSites 1000G_phase1.indels.hg19.vcf
-o ChrALL.100.sam.recal.08-1.grp
第二步:利用第一步生成的ChrALL.100.sam.recal.08-1.grp来生成校正后的数据文件,也是以“.grp”命名,这一步主要是为了与校正之前的数据进行比较,最后生成碱基质量值校正前后的比较图,如果不想生成最后BQSR比较图,这一步可以省略。
e.g.
java -jar GenomeAnalysisTK.jar
-T BaseRecalibrator
-R hg19.fa
-I ChrALL.100.sam.dedup.realn.07.bam
-BQSR ChrALL.100.sam.recal.08-1.grp
-o GATK/hg19.recal.08-2.grp
-knownSites dbsnp_137.hg19.vcf
-knownSites Mills_and_1000G_gold_standard.indels.hg19.vcf
-knownSites 1000G_phase1.indels.hg19.vcf
第三步:利用工具PrintReads将经过质量值校正的数据输出到新的bam文件中,用于后续的变异检测。
e.g.
java -jar GenomeAnalysisTK.jar
-T PrintReads
-R hg19.fa
-I ChrALL.100.sam.dedup.realn.07.bam
-BQSR ChrALL.100.sam.recal.08-1.grp
-o ChrALL.100.sam.recal.08-3.grp.bam
主要参数说明:
-bqsrBAQGOP:BQSR BAQ gap open 罚值,默认值是40,如果是对全基因组数据进行BQSR分析,设置为30会更好。
-lqt: 在计算过程中,该算法所能考虑的reads两端的最小质量值。如果质量值小于该值,计算过程中将不予考虑,默认值是2。
注意:
(1)当bam文件中的reads数量过少时,BQSR可能不会正常工作,GATK网站建议base数量要大于100M才能得到比较好的结果。
(2)除非你所研究的样本所得到的reads数实在太少,或者比对结果中的mismatch基本上都是实际存在的变异,否则必须要进行BQSR这一步。对于人类基因组,即使有了dbSNP和千人基因组的数据,还有很多mismatch是错误的。因此,这一步能做一定要做。
step 9: 分析和评估BQSR结果
这一步会生成评估前后碱基质量值的比较结果,可以选择使用图片和表格的形式展示。
e.g.
java -jar GenomeAnalysisTK.jar
-T AnalyzeCovariates
-R hg19.fa
-before ChrALL.100.sam.recal.08-1.grp
-after ChrALL.100.sam.recal.08-2.grp
-csv ChrALL.100.sam.recal.grp.09.csv
-plots ChrALL.100.sam.recal.grp.09.pdf
参数解释:
-before: 基于原始比对结果生成的第一次校对表格。
-after: 基于第一次校对表格生成的第二次校对表格。
-plots: 评估BQSR结果的报告文件。
-csv: 生成报告中图标所需要的所有数据。
step 10: Reduce bam file
这一步是使用ReduceReads这个工具将bam文件进行压缩,生成新的bam文件,新的bam文件仍然保持bam文件的格式和所有进行变异检测所需要的信息。这样不仅能够节省存储空间,也方便后续变异检测过程中对数据的处理。
e.g.
java -jar GenomeAnalysisTK.jar
-T ReduceReads
-R hg19.fa
-I ChrALL.100.sam.recal.08-3.grp.bam
-o ChrALL.100.sam.recal.08-3.grp.reduce.bam
到此为止,GATK流程中的第一大步骤就结束了,完成了variants calling所需要的所有准备工作,生成了用于下一步变异检测的bam文件。
step 11: call snp
来自:https://www.plob.org/article/7023.html
GATK在这一步里面提供了两个工具进行变异检测——UnifiedGenotyper和HaplotypeCaller。其中HaplotypeCaller一直还在开发之中,包括生成的结果以及计算模型和命令行参数一直在变动,因此,目前使用比较多的还是UnifiedGenotyper。此外,HaplotypeCaller不支持Reduce之后的bam文件,因此,当选择使用HaplotypeCaller进行变异检测时,不需要进行Reduce reads。
UnifiedGenotyper是集合多种变异检测方法而成的一种Variants Caller,既可以用于单个样本的变异检测,也可以用于群体的变异检测。UnifiedGenotyper使用贝叶斯最大似然模型,同时估计基因型和基因频率,最后对每一个样本的每一个变异位点和基因型都会给出一个精确的后验概率。
e.g.
java -jar GenomeAnalysisTK.jar
-glm BOTH
-l INFO
-R hg19.fa
-T UnifiedGenotyper
-I ChrALL.100.sam.recal.08-3.grp.reduce.bam
-D dbsnp_137.hg19.vcf
-o ChrALL.100.sam.recal.10.vcf
-metrics ChrALL.100.sam.recal.10.metrics
-stand_call_conf 10
-stand_emit_conf 30
上述命令将对输入的bam文件中的所有样本进行变异检测,最后生成一个vcf文件,vcf文件中会包含所有样本的变异位点和基因型信息。但是现在所得到的结果是最原始的、没有经过任何过滤和校正的Variants集合。这一步产生的变异位点会有很高的假阳性,尤其是indel,因此,必须要进行进一步的筛选过滤。这一步还可以指定对基因组的某一区域进行变异检测,只需要增加一个参数 -L:target_interval.list,格式是bed格式文件。
主要参数解释:
-A: 指定一个或者多个注释信息,最后输出到vcf文件中。
-XA: 指定不做哪些注释,最后不会输出到vcf文件中。
-D: 已知的snp文件。
-glm: 选择检测变异的类型。SNP表示只进行snp检测;INDEL表示只对indel进行检测;BOTH表示同时检测snp和indel。默认值是SNP。
-hets: 杂合度的值,用于计算先验概率。默认值是0.001。
-maxAltAlleles: 容许存在的最大alt allele的数目,默认值是6。这个参数要特别注意,不要轻易修改默认值,程序设置的默认值几乎可以满足所有的分析,如果修改了可能会导致程序无法运行。
-mbq: 变异检测时,碱基的最小质量值。如果小于这个值,将不会对其进行变异检测。这个参数不适用于indel检测,默认值是17。
-minIndelCnt: 在做indel calling的时候,支持一个indel的最少read数量。也就是说,如果同时有多少条reads同时支持一个候选indel时,软件才开始进行indel calling。降低这个值可以增加indel calling的敏感度,但是会增加耗费的时间和假阳性。
-minIndelFrac: 在做indel calling的时候,支持一个indel的reads数量占比对到该indel位置的所有reads数量的百分比。也就是说,只有同时满足-minIndelCnt和-minIndelFrac两个参数条件时,才会进行indel calling。
-onlyEmitSamples:当指定这个参数时,只有指定的样本的变异检测结果会输出到vcf文件中。
-stand_emit_conf:在变异检测过程中,所容许的最小质量值。只有大于等于这个设定值的变异位点会被输出到结果中。
-stand_call_conf:在变异检测过程中,用于区分低质量变异位点和高质量变异位点的阈值。只有质量值高于这个阈值的位点才会被视为高质量的。低于这个质量值的变异位点会在输出结果中标注LowQual。在千人基因组计划第二阶段的变异检测时,利用35x的数据进行snp calling的时候,当设置成50时,有大概10%的假阳性。
-dcov: 这个参数用于控制检测变异数据的coverage(X),4X的数据可以设置为40,大于30X的数据可以设置为200。
注意:GATK进行变异检测的时候,是按照染色体排序顺序进行的(先call chr1,然后chr2,然后chr3…最后chrY),并非多条染色体并行检测的,因此,如果数据量比较大的话,建议分染色体分别进行,对性染色体的变异检测可以同常染色体方法。
大多数参数的默认值可以满足大多数研究的需求,因此,在做变异检测过程中,如果对参数意义不是很明确,不建议修改。
实验室常用脚本中,-T UnifiedGenotyper 后面会加几个参数:
-T UnifiedGenotyper -nt 6 -stand_call_conf 30.0 -stand_emit_conf 30.0 -rf MappingQuality -mmq 20
-nt: 线程数
-stand_call_conf 30.0: --standard_min_confidence_threshold_for_calling double default: 30.0
The minimum phred-scaled confidence threshold at which variants should be called. The minimum phred-scaled Qscore threshold to separate high confidence from low confidence calls. Only genotypes with confidence >= this threshold are emitted as called sites. A reasonable threshold is 30 for high-pass calling (this is the default).
-stand_emit_conf 30.0: --standard_min_confidence_threshold_for_emitting double default: 30.0
The minimum phred-scaled confidence threshold at which variants should be emitted (and filtered with LowQual if less than the calling threshold). This argument allows you to emit low quality calls as filtered records. http://www.chenlianfu.com/?p=1523
-rf MappingQuality -mmq 20: GATK提供的CountReads工具中,可以过滤read
功能: 计算reads数
分类: 诊断和质量控制工具
概要: 最好和–read-filter合用,这样可以了解下符合特定标准的reads数
输入: 一个或多个BAM文件
输出: 结果会输出到屏幕(标准输出)上,毕竟是用来确定阈值的,也不需要一定要输出到文件中
e.g.:
java -jar ~/biosoft/GenomeAnalysisTK.jar -T CountReads -R $work/database/TAIR10/TAIR10.fa -I BC_bg_reads.sorted.bam
输出:
CountReads - CountReads counted 55080781 reads in the traversal
–read-filter/-rf后面可以接很多的选项,官方文档列出了如下内容:(参考:https://www.jianshu.com/p/710c51dcf1be)
这里主要用到-rf MappingQuality,Filter out reads with low mapping qualities
java -jar GenomeAnalysisTk.jar \
-T HaplotypeCaller \
-R reference.fasta \
-I input.bam \
-o output.vcf \
-rf MappingQuality \
-mmq 15
用HaplotypeCaller call snp运行方法与 UnifiedGenotyper大同小异,至此,从原始.fq.gz文件到获得变异位点的WGS处理就完成了。
若想要call indel,方法有很多,可以参考这篇博文:http://bioinformatics.lofter.com/post/bffd5_a87dee
这里粘贴用 GATK UnifiedGenotyper call indel的方法,只是在call snp的脚本里改了一些参数:
java -jar GenomeAnalysisTKLite.jar
-R ref.fasta
-T UnifiedGenotyper
-I sample.bam
-o sample.gatk.vcf
-nt 4
-stand_call_conf 50.0
-stand_emit_conf 0
-glm INDEL
-rf BadCigar
这边主要是把-glm这个参数设成了INDEL,所以输出的结果当中只有INDEL。
-nt这个参数设的是线程数,除了这个参数还有-nct也可以控制线程数,data threads是个什么概念楼主也比较费解,-nt是分配多少个data threads,而-nct是每个data threads分配多少个CPU,大家看机器资源试着设好了,我一般都用-nt,耗内存高一点不过应该貌似快一点……
然后根据个人经历2.2版本Call出来的Indel会莫名其妙的少掉很多,很多2.1 Call的出来的我用2.2试了恩个参数也Call不出来(肯定是我打开软件的方式不对= =),而且2.2的AD值貌似有bug,明显很多不对,不过-maxAltAlleles默认值已经升到了6而且升值不减速确实很imba(就是这么多alt偶尔会觉得没什么意义的就是……) 不过如果真是bug就有望被修复,坐等GATK继续越做越好。
最后再提一下-rf这个参数,全称是–read_filter,就是用来筛选输入的bam文件中的reads的,因为GATK会检查bam文件里面有个叫Cigar值的东西,有时候有的mapping软件生成的bam文件当中有一些不符合它的标准,在用GATK处理时就可能会包Malformed read一类的错,所以可以通过-rf BadCigar这个参数来剔除掉这些不规范的reads,这样GATK就能正常运行了,上次有同学就碰到这样的问题,我后来才想起来加上这个参数应该大部分相关问题都能解决(如果加上了还不能解决的话那就可能是版本的bug了,GATK的论坛上貌似就有人碰到过这种情况,多换几个版本试试吧……)。
这篇博文中还介绍了其他工具call indel的方法,仅供参考http://bioinformatics.lofter.com/post/bffd5_a87dee。
明天再来介绍call snp之后对原始变异位点进行过滤,并对本片文章之前的步骤进行总结,see you later.